Abstract
Cowpea is an annual legume crop; although it is an essential food in developing countries, cowpea is now grown worldwide. For the genetic improvement of plants, flowering time is one of the major selection criteria. In general, flowering is regulated by photoperiod and temperature, along with the interaction between environmental factors. In this study, we aimed to investigate the candidate genes associated with flowering time using genome-wide association study (GWAS). To investigate the flowering time-related genes, 384 cowpea germplasms were genotyped with 51,128 single nucleotide polymorphisms (SNPs). The main genetic component of days to flowering (DTF) was analyzed using genome association and prediction integrated tool (GAPIT) and elastic-net analyses. From the GAPIT and elastic-net analyses, a total of 23 SNPs were significantly associated with DTF among five (chr. 2, 3, 7, 9, and 11) and seven (chr. 1, 2, 3, 4, 5, 8, and 9) different chromosomes, respectively. Based on our analysis, Vigun01g084000, Vigun01g227200, Vigun02g062600, and Vigun03g296800 were considered the major candidate genes that were significantly associated with DTF in cowpea. These results confirmed that DTF might be controlled by multiple genes affecting early flowering, delaying flowering time, repressing the transition to flowering, etc. This study will potentially contribute to effective DTF genomic selection in plant breeding to better understand the genetic basis and explore the mechanism of flowering time.
-
Key words: Cowpea, Flowering time, SNPs, GWAS, GAPIT, Elastic-net
INTRODUCTION
Cowpea is an annual grain legume that is well-adapted to various environmental conditions such as severe drought, extreme heat, and low soil fertility. However, cowpea is vulnerable to high moisture, chilling stress, and frost (
Elowad and Hall 1987;
Roberts et al. 1993;
Ehlers and Hall 1997;
Coulibaly et al. 2002). Cowpea is a cheap nutritional source of high-quality protein and vitamins, commonly grown in Sub-Saharan Africa and tropical lands (
Ehlers and Hall 1997;
Coulibaly et al. 2002;
Lonardi et al. 2019). However, cowpea is not just an essential food in African or developing countries; it is currently grown worldwide (
Perrino et al. 1993;
Carvalho et al. 2017). For the genetic improvement of plants, flowering time is one of the major selection criteria (
Kim et al. 2012;
Andargie et al. 2013). In addition, it is the essential component for the adoption (
Roberts et al. 1993;
Ishiyaku et al. 2005). Flowering has been commonly regulated by photoperiod and temperature, as well as the interaction between environmental factors (
Uzun 2006;
Andargie et al. 2013). Many genes that control flowering time have been identified in several plant species. Plant photoreceptors, including phytochromes and cryptochromes, allow plants to perceive the day length.
Arabidopsis has five phytochromes genes (
PHYA to
PHYE) and at least two cryptochrome genes (
CRY1 and
CRY2) (
Franklin and Quail 2010). Additionally, photoperiodic time measurement and seasonal flowering time regulation could be achieved by circadian clock genes, including
ELF3,
TOC1,
CCA1,
LHY, and
GI. Another flowering transition was modulated by
FLOWERING LOCUS T (
FT)/
TERMINAL FLOWER 1 (
TFL1) gene family that encodes floral inducers (florigens) and inhibitors (anti-florigens), respectively (
Song et al. 2015).
Cowpea (640.6 Mbp, 2
n = 22) is a warm-season annual legume and short-day plant (
Lonardi et al. 2019). Previous genetic studies were conducted to identify the quantitative trait loci (QTLs) associated with flowering time in cowpea. QTLs for domestication-related traits, including days to flowering (DTF), were identified from two genetic populations in yardlong bean (
Kongjaimun et al. 2012). Using 159 F7 recombinant inbred lines (RILs) derived from the cross between the cultivated variety 524B and wild variety 219-01,
Andargie et al. (2013) also reported five QTLs related to the time of flowering and three QTLs related to DTF on the genetic linkage map using 202 SSR markers. In asparagus bean, one major QTL for DTF was identified in linkage group 11 (
Xu et al. 2013). Recently,
Lo et al. (2018) constructed a high-density genetic map containing 17,739 single nucleotide polymorphism (SNP) markers and identified two major QTLs associated with DTF on Vu05 and Vu09. Based on fine-mapping, phytochrome E (
Vigun09g050600) and transcription factor TCP 18 (
Vigun09g062200) were proposed as candidate genes for one major QTL on Vu09.
The availability of large-scale sequencing and the development of high-density SNP markers provide higher resolution of QTL mapping than other marker systems (
Li et al. 2008;
Mammadov et al. 2012). In addition, the identification of millions of SNP markers across the genome makes it possible to perform a genome-wide association study (GWAS) (
Xu and Taylor 2009). Unlike genetic mapping using bi-parental populations, GWAS usually uses natural populations to identify the candidate genes or loci of agronomic traits by detecting the association between genotypes and phenotypes of target traits. GWAS depends on linkage disequilibrium (LD), i.e., the non-random association of alleles at two or more loci in a population, between markers and QTL (
Flint-Garcia et al. 2003). It is important to identify genetic markers and narrow down the location of the causal variants (
Varshney et al. 2009;
Xu et al. 2012). Generally, self-pollinating crops have higher LD than cross-pollinating crops; thus, the former contribute less number of markers for GWAS and result in mapping at a lower resolution (
Gaut and Long 2003;
Gupta et al. 2005;
Jaiswal et al. 2019). However, GWAS has several limitations including confounding effect of population structure, requirement of high number of individuals and SNP markers, misleading natural variation, and high false-positive association (
Hiersche et al. 2013;
Visscher et al. 2017).
To improve the statistical power of GWAS, many software programs and methods have been developed. Among the GWAS software programs, TASSEL (trait analysis by association, evolution, and linkage) has been commonly used for plants (
Garcia et al. 2019;
Alqudah et al. 2020).
Tang et al. (2016) developed an R-based package called genome association and prediction integrated tool (GAPIT version 2). GAPIT packages contain mixed linear model (MLM), compressed MLM (CMLM) (
Lipka et al. 2012), and enriched CMLM (ECMLM;
Tang et al. 2016). MLM controls the false positives and incorporates both population structure and random effects relationship (
Yu et al. 2006), whereas the ECMLM uses large data samples by clustering individuals into groups and is, therefore, a more efficient and powerful model (
Zhang et al. 2010). Another statistical method, elastic-net, combines the strength of both the least absolute shrinkage and selection operator (LASSO) and ridge regression best linear unbiased prediction (RR-BLUP) (
Wimmer et al. 2013). It is performed with large variables, then groups and shrinks the parameters associated with the correlated variables, and leaves them in the equation or removes them all at once (
Waldmann et al. 2013).
In this study, we investigated SNP loci associated with the flowering time in Korean cowpea germplasm and determined the candidate genes. The result will contribute to the development of new cowpea cultivars in Korea.
MATERIALS AND METHODS
Plant materials and phenotypic evaluation
A total of 384 cowpea accessions were obtained from the Jeonnam Agricultural Research & Extension Services and the Rural Development Administration (RDA) Genebank at the National Agrobiodiversity Center, Republic of Korea. The accessions originated from Benin (1), Bulgaria (7), Cambodia (1), Cote d’Ivoire (1), China (5), Ghana (1), Guyana (1), India (1), Japan (1), Korea (243), Kyrgyzstan (2), Mozambique (1), Myanmar (2), Nepal (3), Nigeria (55), Pakistan (1), Philippines (5), Sri Lanka (2), Taiwan (1), Thailand (5), Uzbekistan (6), Vietnam (4), and unknown (35) (
Supplementary Table S1). The cowpea accessions were planted in the experimental field of Chonnam National University on June 22, 2018 and June 19, 2019. Each accession was planted on 1 m × 1 m hill plots with one replicate. The DTF was recorded in the field by subtracting the planting date from the date when two to three flowers bloomed in each individual. We examined the phenotypes in 2018 and 2019 and produced three different phenotype data by calculation: 2018, 2019, and the average of each year.
Fresh leaves were sampled from each of the 384 individuals. The genomic DNA was isolated from about 150-200 mg of leaf tissue from each accession using a CTAB based on a modified form of method described by
Keim et al. (1988). To verify DNA quality and concentrations, gel electrophoresis and Nano Drop 2000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA) were used. After DNA isolation, 384 cowpea accessions were genotyped with 51K Cowpea iSelect consortium array at TNT Research Co. (Anyang, Gyeonggi-do, Republic of Korea) (
Lonardi et al. 2019;
Seo et al. 2020). SNP calling was conducted with GenomeStudio v.2.0 software (Illumina Inc., CA, USA) using the same custom cluster file as in
Lo et al. (2018). The raw data were filtered for quality control. First, if there was any missing value in the phenotype, the corresponding sample was also removed. Second, the SNPs with minor allele frequency (MAF) < 0.01 and call rate < 95% were eliminated. Third, missing genotypes were imputed from the distributions of allele frequency after calculating MAF from an R package “synbreed” (
Wimmer et al. 2012). Additionally, we transformed the flowering time such that it follows a normal distribution.
The LD values were calculated using the sample correlation coefficient with a sliding window technique of size 400 in the first chromosome. This was similar to GAPIT, where the values were calculated for all pairs of adjacent 400 SNPs with moving starting point.
GWAS analysis
We conducted two types of GWAS: GAPIT and elastic-net model. In GAPIT, ECMLM analysis was performed (
Tang et al. 2016), in which the optimal number of clusters was chosen by comparing the Bayesian Information Criterion (BIC) values for each number of clusters chosen. ECMLM considers the population structure using a kinship matrix. In this study, VanRaden method (VanRaden 2008) was used to estimate a kinship matrix.
GAPIT calculated the P-values of each SNP, and the significance threshold of P-value was calculated using the Bonferroni correction to adjust the multiple testing problem.
The elastic-net model (
Zou and Hastie 2005) is widely used to estimate regression coefficients simultaneously using a penalty function that combines both the lasso and ridge penalties. Generally, the lasso performs the variable selection using sparsity-inducing penalty with less correlated variables, while the ridge controls the smoothness when variables are highly correlated. The elastic-net model selects variables even when there are correlated variables by taking advantage of both the lasso and ridge penalties. This model depends on two tuning parameters: α, which is a mixing proportion between two penalties, and l, which governs how much penalty will be imposed on the parameter estimator.
Based on resampling approach,
Meinshausen and Bühlmann (2010) proposed the selection probability that not only prioritizes the significances between SNPs and phenotypes but also does not depend on the tuning parameters.
Kim et al. (2020) proposed the empirical threshold for the selection probability that can control the number of falsely selected variables.
In this study, SNPs were selected by both Bonferroni-adjusted P-values of GAPIT and selection probabilities of elastic-net model based on empirical threshold.
Prediction of candidate genes for DTF
Candidate genes associated with DTF were searched at around 200 kb flanking to the most significant SNP loci. The predicted gene models were used in
Vigna unguiculata v1.1 on phytozome (
www.phytozome-next.jgi.doe.gov) (
Lonardi et al. 2019). Through functional similarity with other species, such as
Arabidopsis, rice, and legume, candidate genes were identified.
RESULTS
Frequency distribution of DTF in Korean cowpea germplasm
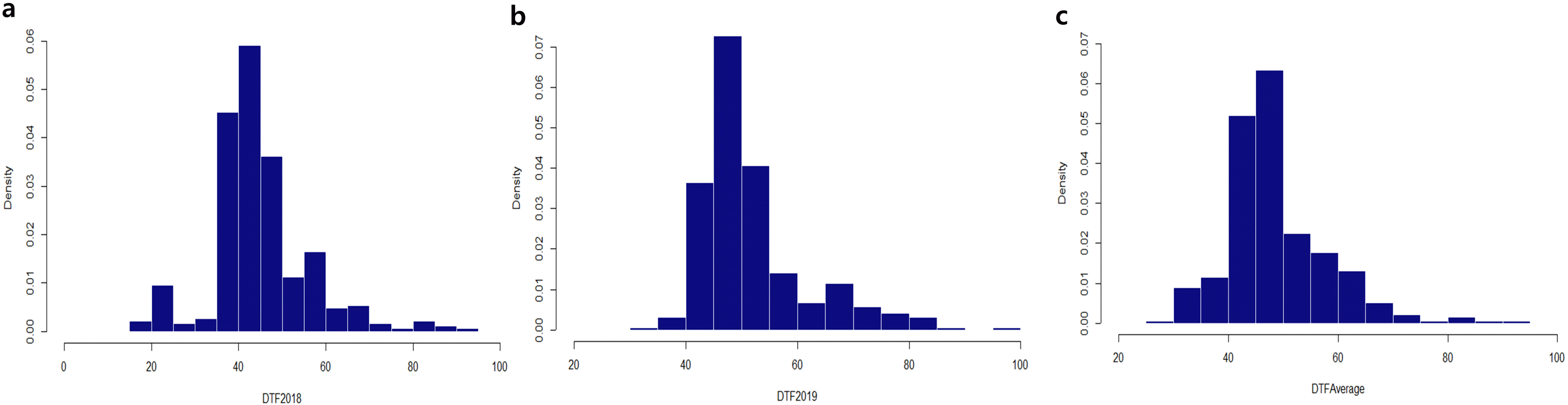
The DTF were measured in 384 cowpea accessions in 2018 and 2019. The skewed frequency distribution was positive, and kurtosis appeared sharply in approximately 40-50 days. However, the overall average of DTF appears to show a similar trend regardless of environment factors (
Table 1 and
Fig. 1). In 2018, DTF ranged from 20 to 93 days, with an average of 45.3 days. Four cowpea accessions (Vu_104, Vu_119, Vu_149, and Vu_154) showed early flowering (
Supplementary Table S1). The Vu_253 accession from Korea showed the greatest delay in flowering time (93 days). In 2019, DTF ranged from 35 (Vu_149) to 97 days (Vu_315), with an average of 52.5 days. From the overall average of DTF measurements, the Vu_149 accession bloomed the fastest: 2018 (20 days), 2019 (40 days), and average (30 days).
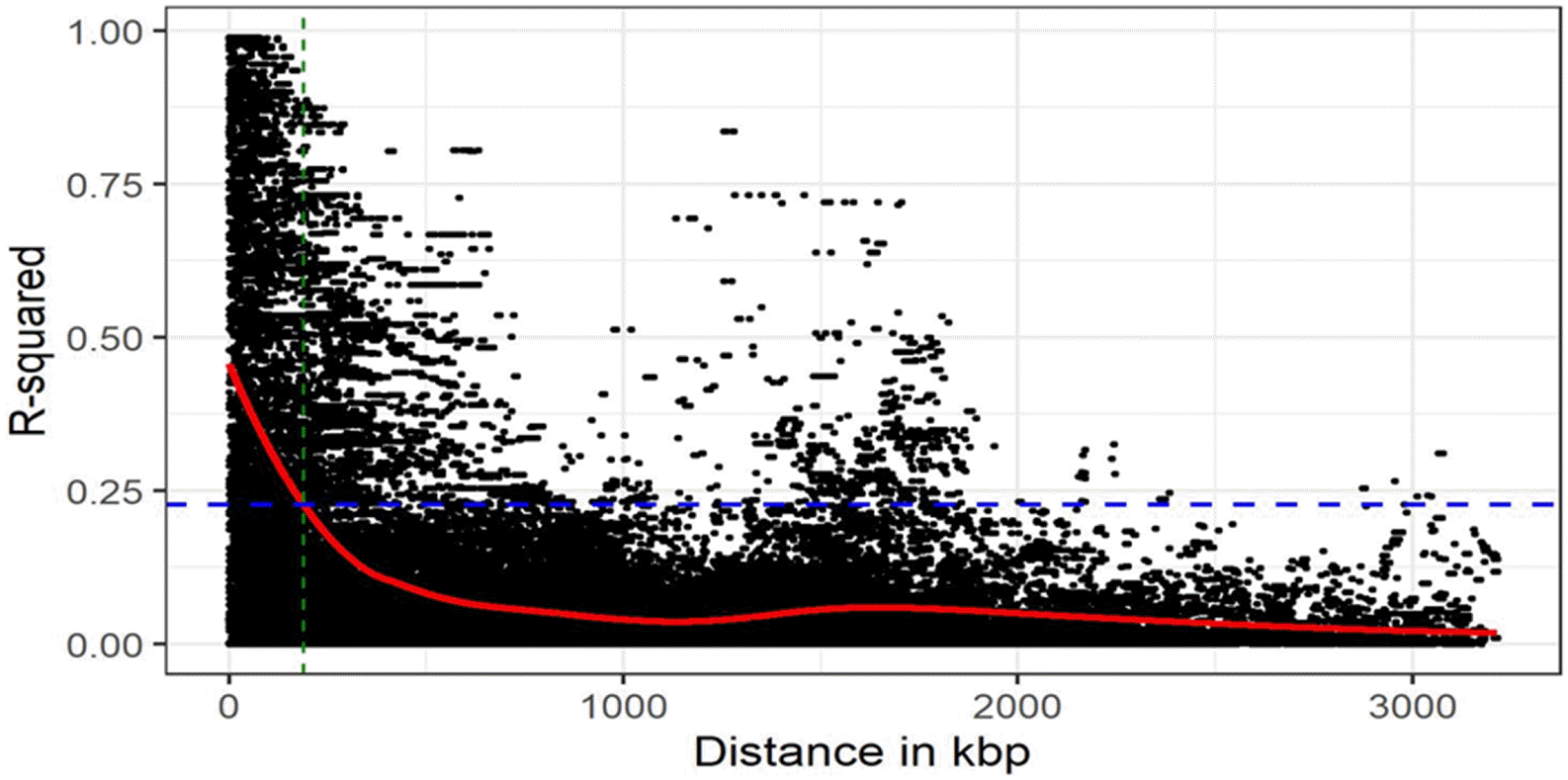
To genotype 384 cowpea accessions, we used Cowpea iSelect Consortium Array (Illunina, Inc.) containing 51,128 SNPs. After removing the markers with MAF < 0.01 and call rate < 95%, a total of 35,600 SNPs remained. These filtered SNP markers provided an average marker density of 1 SNP every 13.5 kb genome-wide, varying across chromosomes, from 10.3 kb/SNP on chr. 7 to 16.8 kb/SNP on chr. 4. With an LD window of 400, genome-wide LD decay was estimated at approximately 188 kb, where
r2 = 0.23 (half of its maximum value
r2 = 0.46;
Fig. 2).
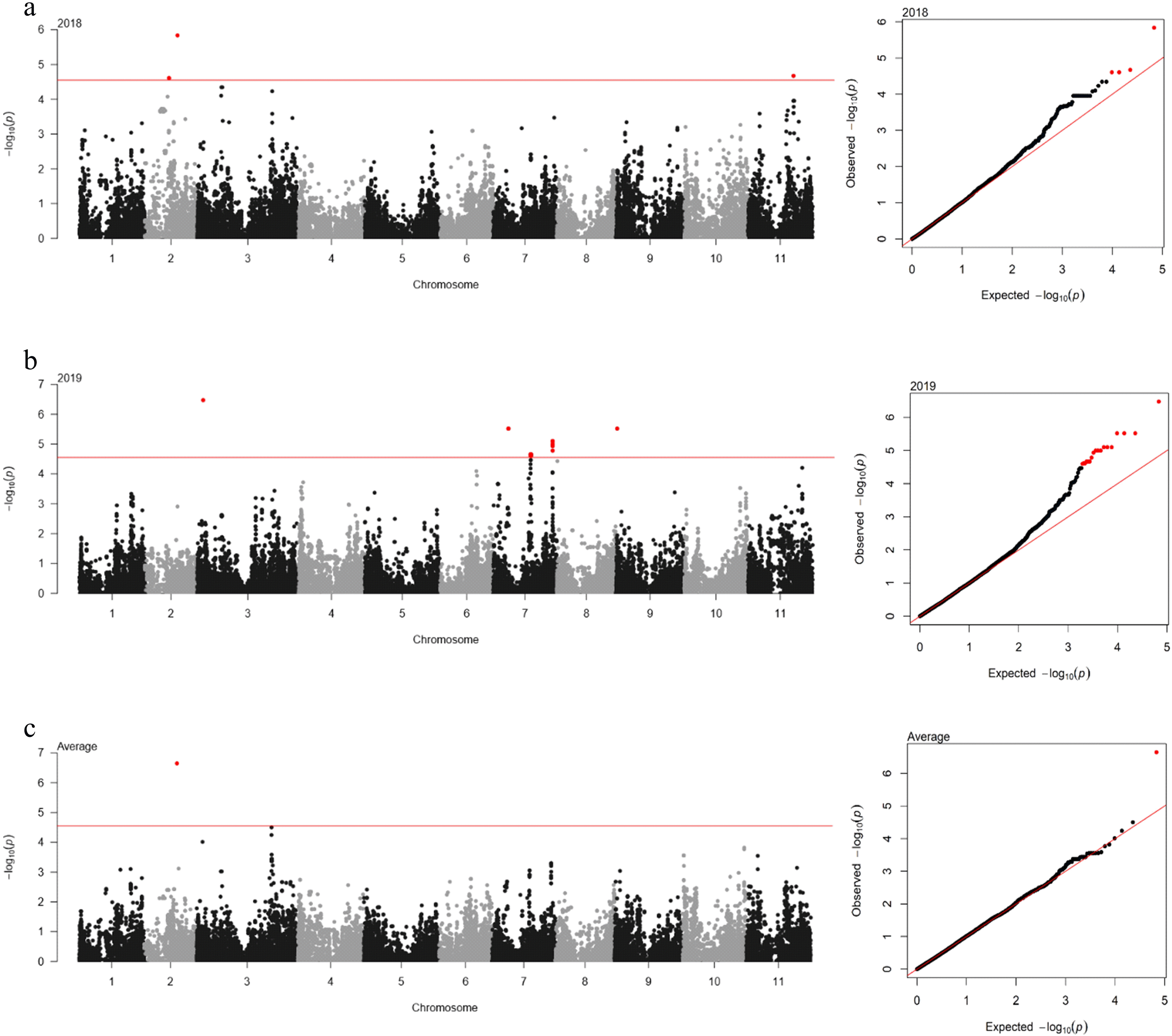
The association between 35,600 SNP markers and DTF was analyzed using ECMLM. The significant cut-off was set at ‒log(
P-value) = 4.55 using the Bonferroni method. A total of 23 SNP loci significantly associated with DTF were identified across years (
Table 2 and
Fig. 3). In 2018, four SNP loci were detected on three different genomic regions on chromosomes 2 and 11. The 2_06981 SNP locus on chromosome 2 showed the highest phenotypic variation (4.7%). In 2019, 18 SNP loci were identified on five genomic regions on chromosomes 3, 7, and 9. The 2_07930 SNP locus on chromosome 3 had the highest phenotypic variation (4.3%). However, only one SNP locus (2_06981) on chromosome 2 was identified for the average of each year.
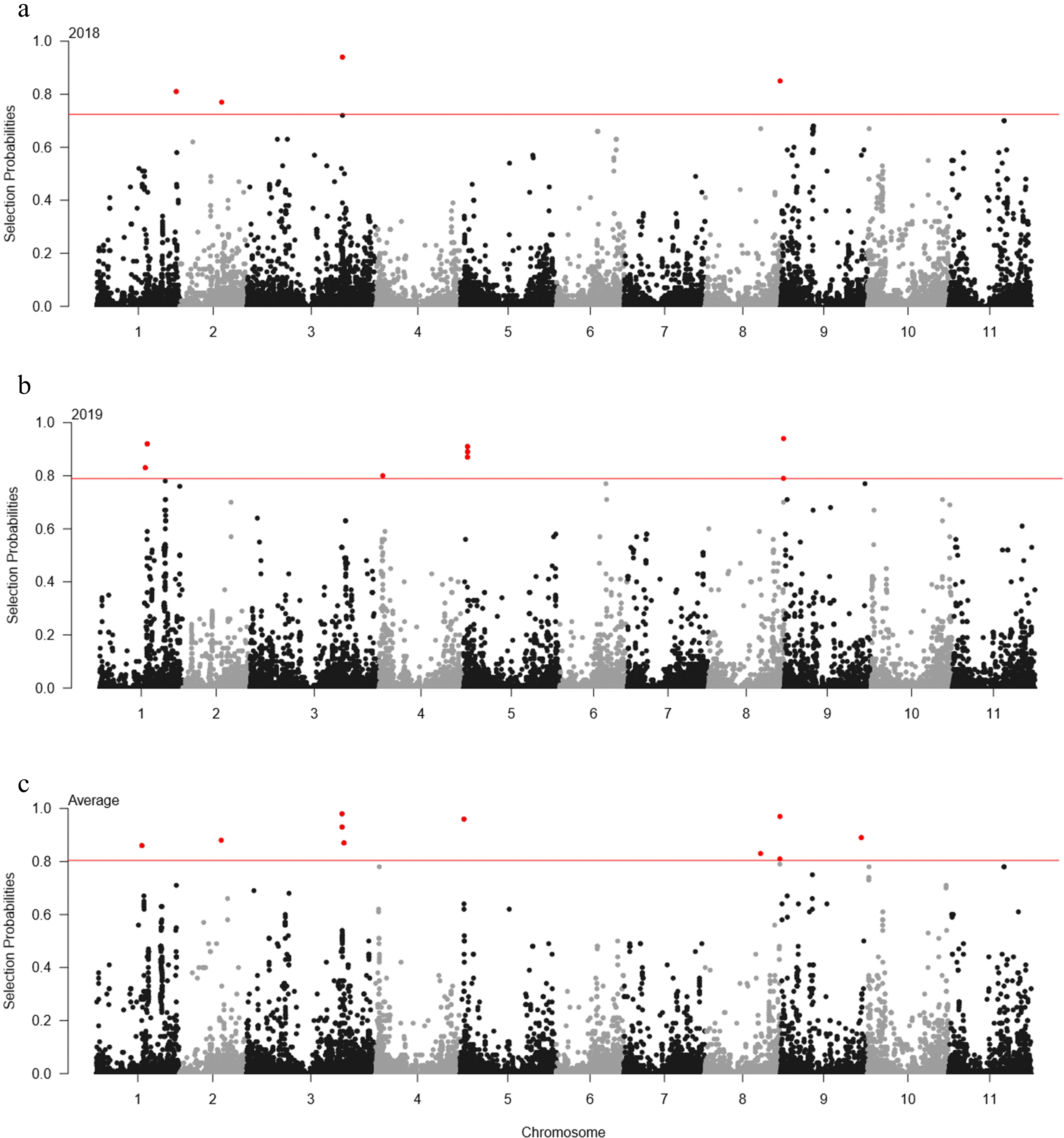
For elastic-net model, the significant cut-offs for 2018, 2019, and the average of each year were set at probability > 0.72, 0.79, and 0.80, respectively. In 2018, five SNP loci were detected in chromosomes 1, 2, 3, and 8 (
Table 3 and
Fig. 4). In 2019, eight SNP loci were identified in chromosomes 1, 4, 5, and 8. On average, 10 SNP loci were identified across six chromosomes, including chromosomes 1, 2, 3, 5, 8, and 9. The 2_54082 SNP locus on chromosome 8 was detected across 2018, 2019, and the average of each year.
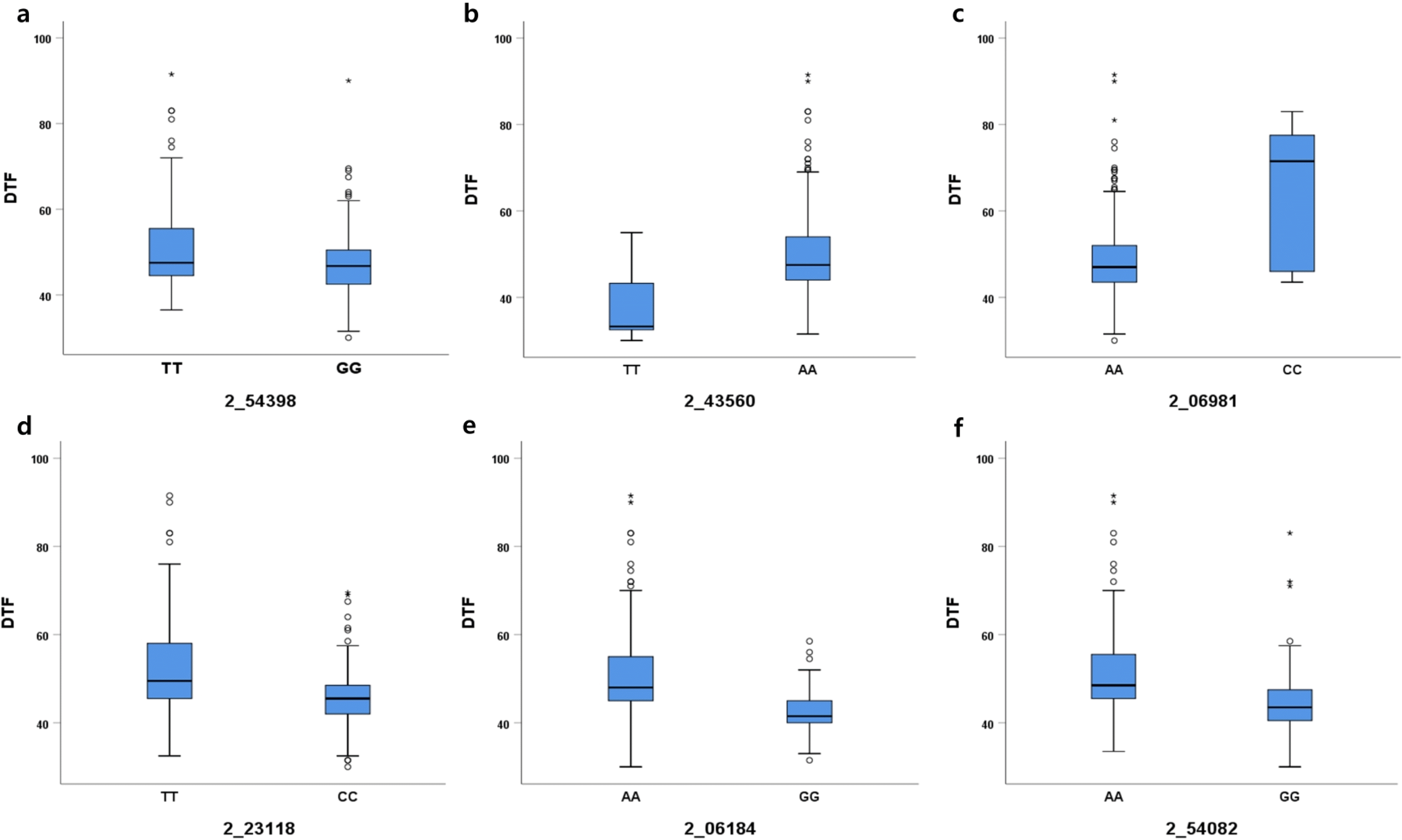
To verify the effects of the associated SNPs on DTF, the distribution of average DTF was examined based on each SNP allele (
Fig. 5). In 2_54398 SNP locus on chromosome 1, 173 cowpea accessions containing the TT allele had 51.1 DTF, and 210 accessions containing the TT allele had 47.1 DTF. In 2_43560 SNP locus, 20 cowpea accessions containing the TT allele had 37.3 DTF and 362 accessions containing the AA allele had 49.6 DTF. The 2_06981 locus on chromosome 2 showed the largest difference (16 days) in DTF between AA (48.6 DTF) and CC alleles (64.6 DTF). In addition, 2_23118 on chromosome 3, 2_06184 on chromosome 5, and 2_54082 on chromosome 8 showed 6.6 days, 8.0 days, and 7.4 days of differences in DTF, respectively, between lines carrying different alleles of the SNPs. The results indicated the noticeable differences in DTF, which may be affected by alleles.
From the GWAS with ECMLM and elastic-net, six significantly common regions were detected on chromosomes 1, 2, 3, 5, and 8. A total of 22 flowering-related genes were identified from the interval of 200 kb on either side of significant SNPs (
Table 4). Among them, many genes related to flowering time regulation (10), expression in flowering (7), and others (5) were found. Especially, FLOWERING LOCUS T (
Vigun01g084000) and EARLY FLOWERING 4 (
Vigun01g227200) were located on chromosome 1, and F-BOX/KELCH-REPEAT PROTEIN SKIP11 (
Vigun02g062600) was located on chromosome 2. On chromosome 3, Casein kinase II (
Vigun03g296800) was identified. These genes are mainly involved in the photoperiod response pathway, floral organ formation, flower and leaf senescence, etc. In addition, other genes are also closely associated with the control of flowering time, such as delayed flowering time, flower development, flowering signals, and repression of the transition to flowering.
DISCUSSION
Flowering time has a major influence on plant growth that allow improvement in crop yield (
Amasino and Michaels 2010;
Andargie et al. 2013). In this study, we measured the DTF of 384 cowpea germplasm including 243 Korean accessions. Most Korean cowpea accessions had approximately 40-50 DTFs, with the lowest of 20 days and the highest of 97 days (
Table 1). These differences in DTF were probably due to the genetic diversity in Korean cowpea accessions. Previously,
Seo et al. (2020) identified that Korean cowpea accessions had been introduced from two different routes, West Africa or Europe, and had similar genetic diversity with the global cowpea accessions. In addition, there were differences in DTF in each province of Korea: Jeolla (49.3 days), Chungcheong (54.3 days), Gangwon (47.5 days), Gyeongsang (52.1 days), Gyeonggi (49.5 days), and Jeju (63.5 days). Jeju had the longest DTF among the provinces. These great phenotypic variability and genetic diversity in this population could provide useful insights into the genetic basis of DTF in cowpea.
GWAS based on the combination of high-throughput SNPs and diverse phenotypes has been used to elucidate the genetic basis of numerous flowering-related genes in various crops such as
Arabidopsis, rice, maize, and other legume crops. In this study, 22 SNPs significantly associated with DTF were identified across years in the two types of statistical models. In ECMLM, four SNP loci on three different genomic regions on chromosomes 2 and 11, and 18 SNP loci on five genomic regions on chromosomes 3, 7, and 9 were identified for 2018 and 2019, respectively. In addition, one SNP locus (2_06981) on chromosome 2 was identified for the average of each year. Previously,
Lo et al. (2018) identified two significant QTLs (CFt5 and CFt9) related to DTF on chromosomes 5 and 9. One QTL, CFt9, which was linked to 2_03945 (5,449,874 bp on Vu09), was located near the 2_11217 (643,886 bp on Vu09) SNP locus identified in this study. In the elastic-net model, five SNP loci on chromosomes 1, 2, 3, and 8, eight SNP loci on chromosomes 1, 4, 5, and 8, and 10 SNP loci on chromosomes 1, 2, 3 5, 8, and 9 were identified in 2018, 2019, and the average of each year, respectively. Especially, 2_06981 on chromosome 2 was detected by both ECMLM and elastic-net models. The 2_06184 SNP locus (2,070,082 bp on Vu05) was detected in 2019 and average and was a similar region previously reported QTL CFt5 (2_05332, 854,745 bp on Vu05) (
Lo et al. 2018). In addition, 2_54082 SNP (37,954, 268 bp on Vu08) was detected on all three conditions and was stable across different environments and might play a key role to control DTF in different genetic background. This region was overlapped with the position (SSR-6188, ∼33.2 Mb on Vu08) of QTL qdtf1 explaining 18.5% of phenotypic variation (
Andargie et al. 2013). The 2_20601 locus at the 40.8 Mb position on chromosome 9 was located near minor QTL qdtf7 (SSR-7027-4, ∼43.3 Mb on Vu09) explaining 5.7% of the phenotypic variation (
Andargie et al. 2013). Based on these results, the elastic-net model was more powerful in detecting QTL associated with DTF. The elastic-net analysis excelled in identifying many influential genetic variants, which is suitable for selecting variables (
Cho et al. 2010). Previously,
Cho et al. (2009) reported that elastic-net regularization helped to identify large number of correlated SNPs for rheumatoid arthritis. Elastic-net regularization also improved the genomic prediction across multiple populations in maize (
Schulz-Streeck et al. 2012).
We identified the candidate genes using a block of approximately 200 kb in length based on the back and forth of selected SNPs (
Zhao et al. 2017). A total of 22 flowering-related genes were found on five chromosomes (
Table 4). The target 2_54398 SNP locus on chromosome 1 contained four candidate genes associated with flowering time. Among them,
Vigun01g084000 encodes protein FT that promotes the transition for reproductive development and flowering. Diverse FT signaling pathways were involved in encoding a pair of flowering regulators homologous to phosphatidylethanolamine-binding proteins (PEBPs) (
Wickland and Hanzawa 2015;
Liu et al. 2016). Within the LD blocks of 2_43560 on chromosome 1,
Vigun01g227200 encoding PROTEIN EARLY FLOWERING 4 functions as an important key gene involved in the promotion of photoperiod and expression of the control of flowering time (
Khanna et al. 2003). On chromosome 2,
Vigun02g062600 is located 154 kb up-stream of 2_06981. This gene encodes F-BOX/KELCH-REPEAT PROTEIN SKIP11. The kelch containing F-box protein (KFB) is only found together in plant proteins and affects the photoperiodic flowering pathway depending on light signals by controlling the circadian clock and photoperiodic flowering processes (
Xu et al. 2009). The functions of ZEITLUPE (ZTL) and FLAVIN-BINDING KELCH REPEAT F-BOX1 (FKF1) were associated with circadian clock, photomor-phogenesis, and flowering time regulation (
ul Hassan et al. 2015). Among the candidate genes in the region of 2_23118 on chromosome 3,
Vigun03g296800 encoding Casein kinase II regulatory subunit is similar to the circadian clock component; it is one of the protein kinases containing the evolutionarily conserved function (
Ogiso et al. 2010).
In conclusion, we conducted GWAS to identify the SNP loci associated with the DTF using 384 cowpea accessions with 51K SNP array. A total of 23 SNPs were detected using GAPIT and elastic-net analyses. In addition, four major candidate genes were identified for flowering time. These results confirmed that DTF was strongly controlled by multiple genes, affecting early flowering, extending flowering time, suppressing the transition to flowering, etc. This could improve our understanding of the genetic basis and aid in the exploration of the mechanism of flowering time.
SUPPLEMENTARY MATERIALS
ACKNOWLEDGEMENTS
This work was supported by the “Cooperative Research Program for Agriculture Science and Technology Devel-opment (Project No. PJ013125032020)” Rural Development Administration, Republic of Korea.
Fig. 1Distribution of flowering time. (a) 2018; (b) 2019; and (c) average. The horizontal axis indicates the frequency, whereas the vertical axis indicates the days to flowering (DTF).
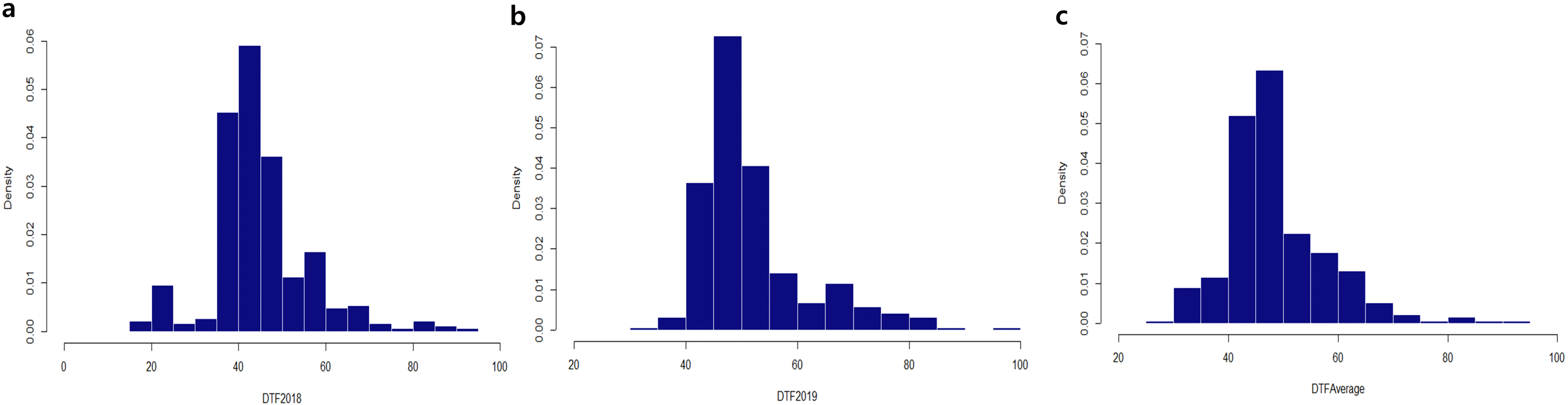
Fig. 2Distribution of marker density and decay of link-age disequilibrium (LD) over distance displayed as an accumulative distribution. The x-axis indicates distance (in kbp) between SNPs, the vertical y-axis indicates LD value (R2), and the blue solid horizontal line indicates the distance of LD pattern in the total genotype of cowpea.
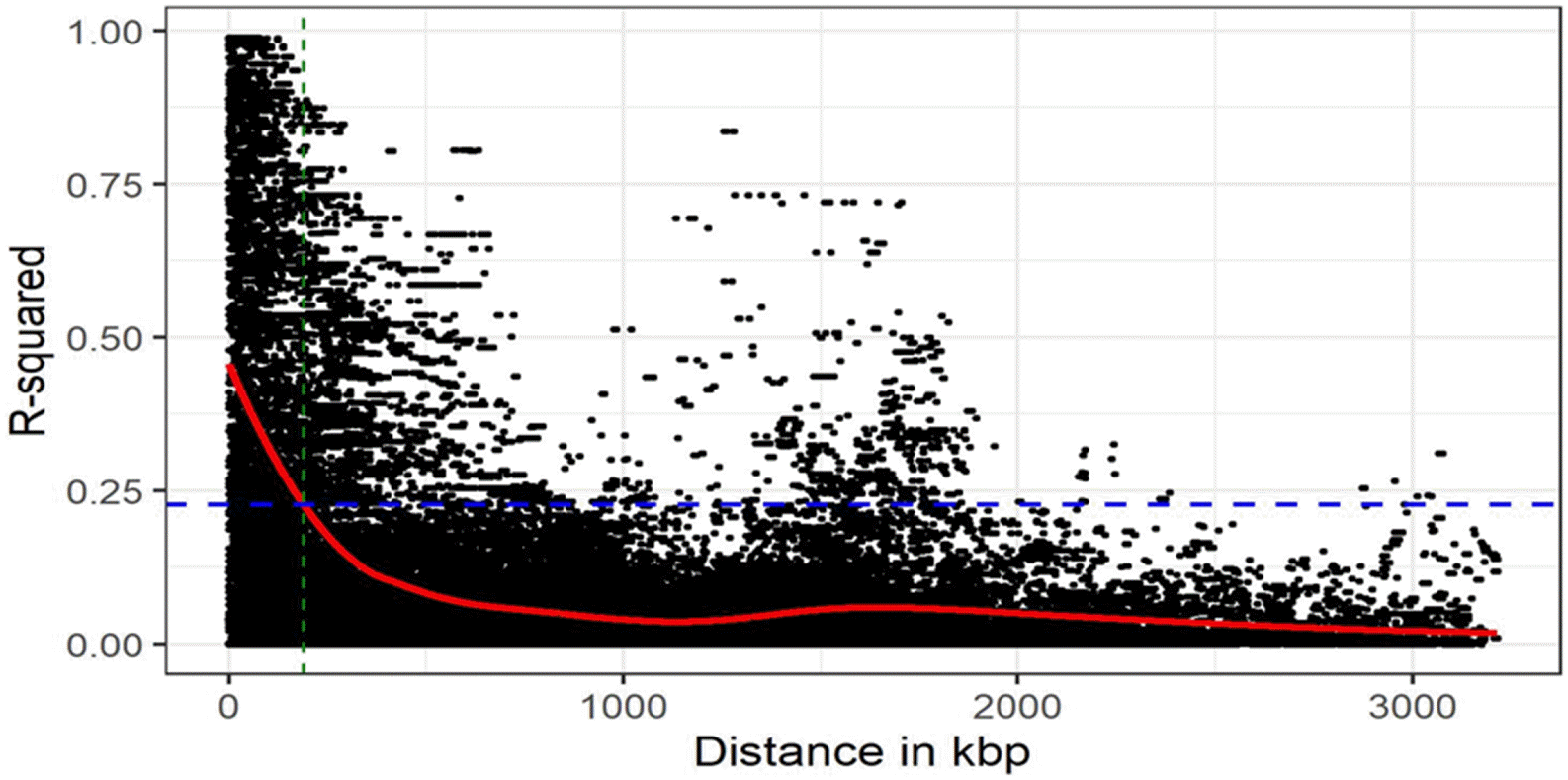
Fig. 3Manhattan plot (left) and quantile-quantile (QQ) plot (right) for the days to flowering (DTF) in ECMLM. (a) 2018; (b) 2019; and (c) average. In Manhattan plot, the horizontal axis indicates the chromosomes; the vertical axis indicates the ‒log10(p); the red solid horizontal line indicates the significance.
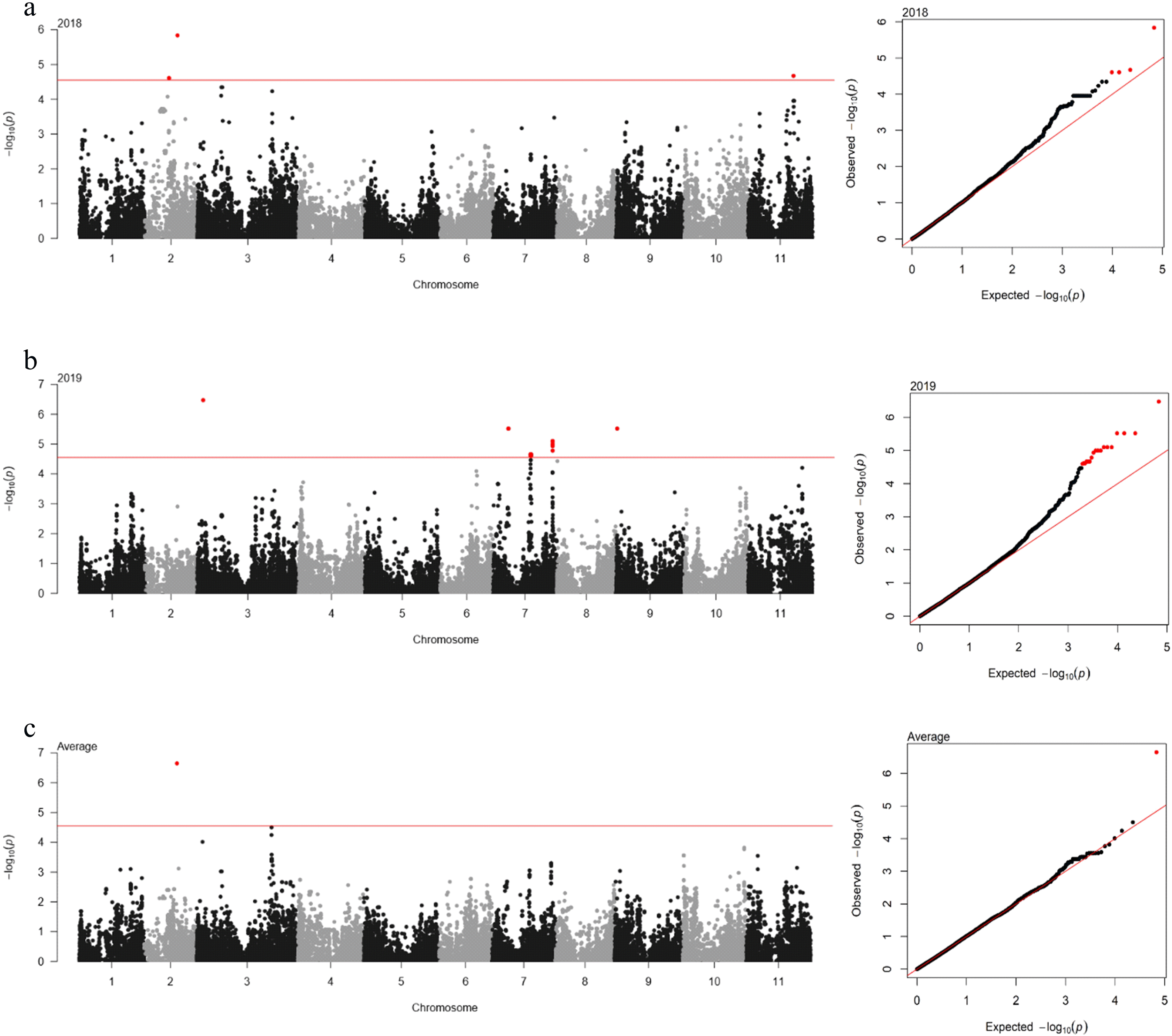
Fig. 4Manhattan plot for the days to flowering (DTF) in elastic-net model. (a) 2018 Manhattan plot; (b) 2019 Manhattan plot; (c) average. In Manhattan plot, the horizontal axis indicates the chromosomes, the vertical axis indicates the selection probability, and the red solid horizontal line indicates the significance.
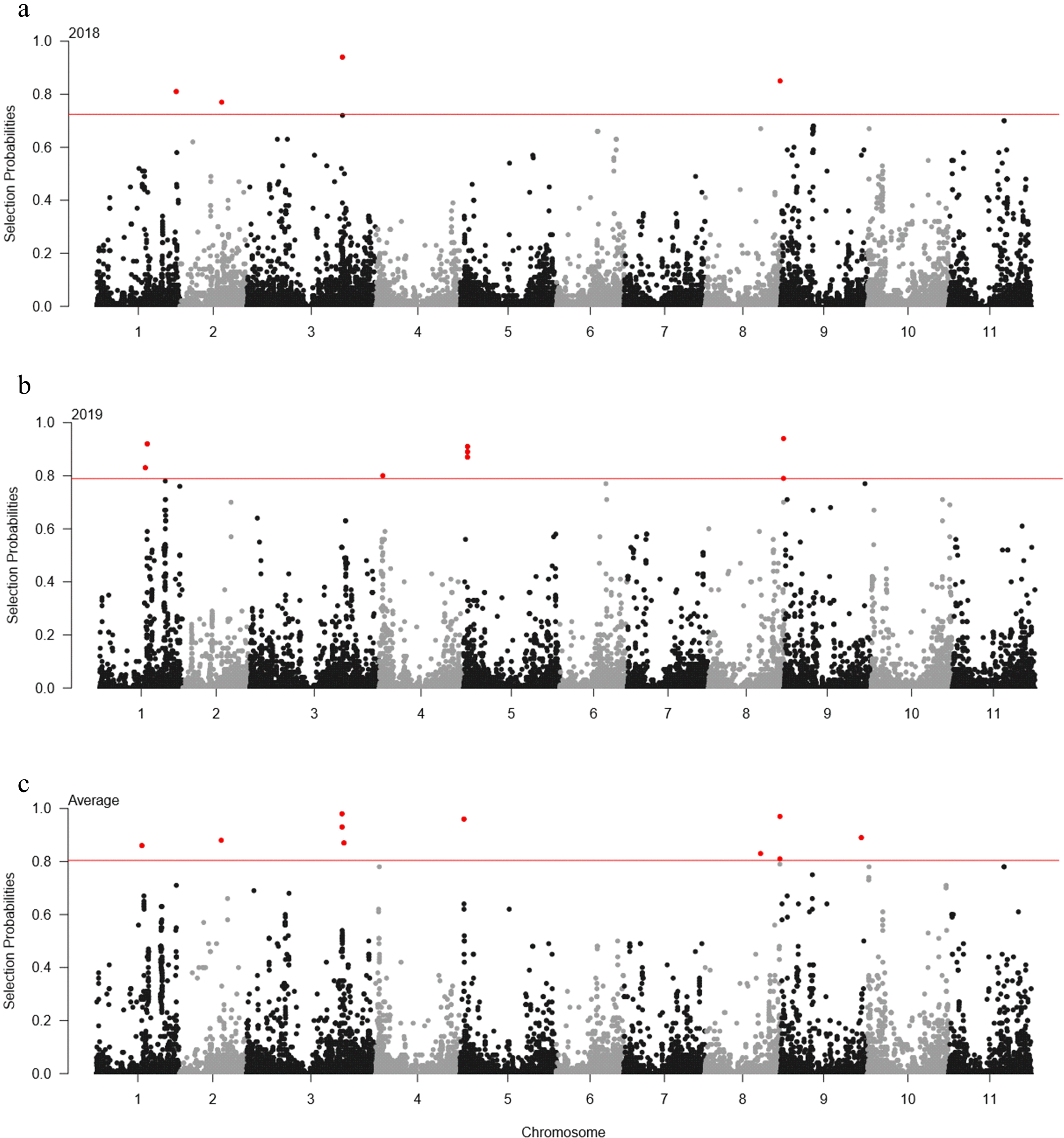
Fig. 5Determination of days to flowering (DTF) represented by box plots. (a) 2_54398 SNP on chromosome 1; (b) 2_43560 SNP on chromosome 1; (c) 2_06981 SNP on chromosome 2; (d) 2_23118 on chromosome 3; (e) 2_06184 SNP on chromosome 5; (f) 2_54082 SNP on chromosome 8.
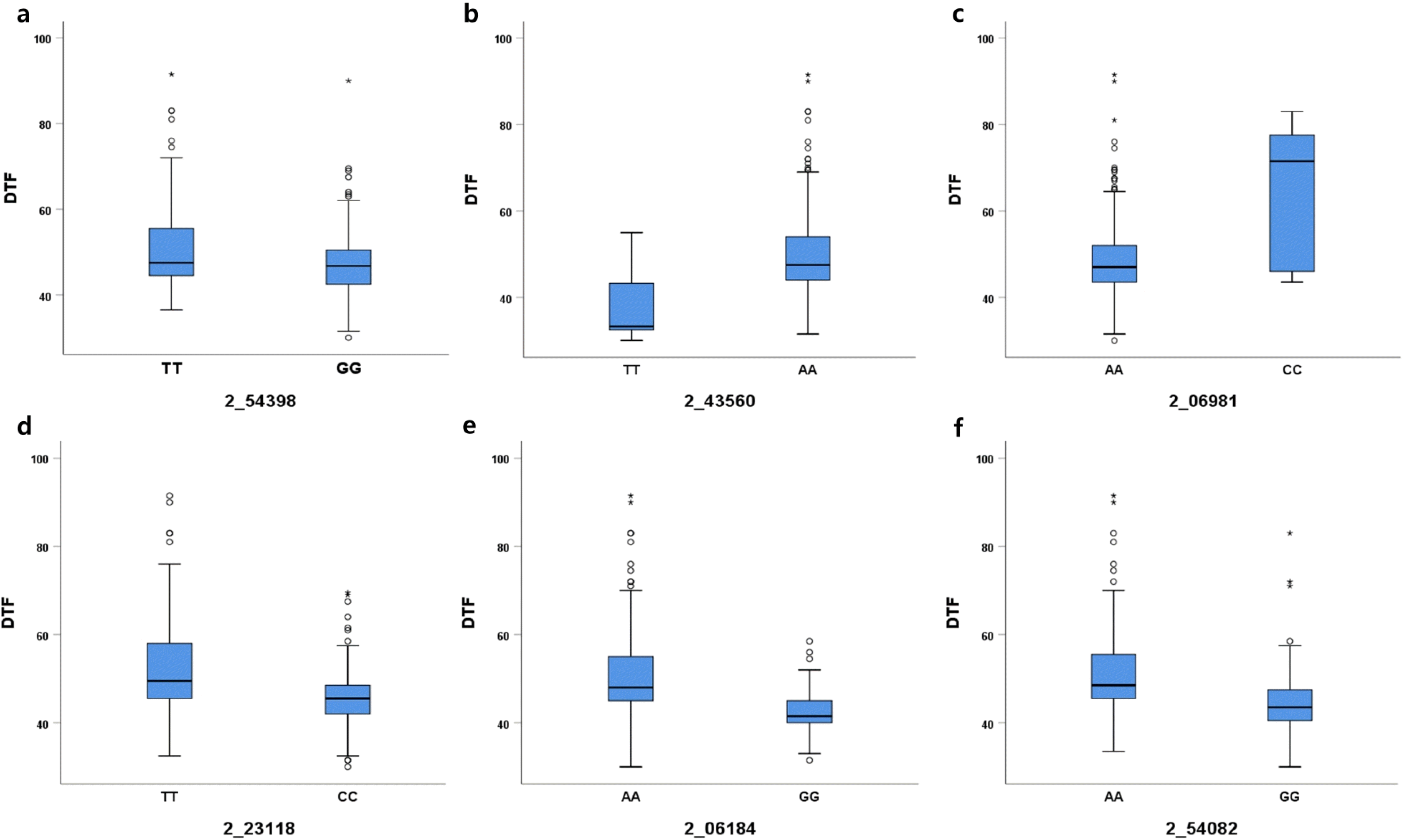
Table 1Statistical analysis of distribution of flowering time.
Table 1
|
Year |
Mean |
SDz)
|
Min |
Max |
Range |
Skewness |
Kurtosis |
|
2018 |
45.3 |
11.4 |
20 |
93 |
73 |
0.95 |
2.72 |
|
2019 |
52.5 |
9.6 |
35 |
97 |
62 |
1.56 |
2.47 |
|
Average |
48.9 |
9.2 |
30 |
91.5 |
61.5 |
1.25 |
2.90 |
Table 2Details of SNP loci associated with flowering time in cowpea identified by genome-wide association study using enrichment of the compressed mixed linear model (ECMLM).
Table 2
|
Year |
SNP marker |
Chr. |
Position (bp) |
P-value |
MAFz)
|
R2y)
|
Effectx)
|
|
2018 |
2_06981 |
2 |
21,120,081 |
1.45E-06 |
0.021 |
0.047 |
10.316 |
|
2_48274 |
2 |
15,616,162 |
2.47E-05 |
0.059 |
0.035 |
‒7.46 |
|
2_27201 |
2 |
15,590,209 |
2.47E-05 |
0.059 |
0.035 |
7.46 |
|
2_18947 |
11 |
29,228,351 |
2.12E-05 |
0.045 |
0.036 |
8.635 |
|
2019 |
2_07930 |
3 |
3,749,050 |
3.00E-07 |
0.03 |
0.043 |
‒7.524 |
|
2_39739 |
7 |
9,599,445 |
3.00E-06 |
0.01 |
0.036 |
‒9.86 |
|
2_49778 |
7 |
9,508,214 |
3.00E-06 |
0.01 |
0.036 |
‒9.86 |
|
2_18350 |
7 |
38,100,144 |
8.00E-06 |
0.458 |
0.033 |
3.955 |
|
2_18351 |
7 |
38,099,882 |
8.00E-06 |
0.458 |
0.033 |
3.955 |
|
2_23266 |
7 |
38,118,029 |
8.00E-06 |
0.458 |
0.033 |
3.955 |
|
2_17728 |
7 |
38,106,355 |
1.00E-05 |
0.455 |
0.032 |
3.896 |
|
2_23267 |
7 |
38,118,317 |
1.00E-05 |
0.455 |
0.032 |
3.896 |
|
2_54832 |
7 |
38,121,357 |
1.00E-05 |
0.455 |
0.032 |
3.896 |
|
2_17729 |
7 |
38,109,623 |
1.00E-05 |
0.461 |
0.032 |
3.875 |
|
2_06047 |
7 |
38,123,502 |
2.00E-05 |
0.455 |
0.031 |
3.784 |
|
2_00153 |
7 |
24,076,659 |
2.00E-05 |
0.058 |
0.03 |
‒6.445 |
|
2_50584 |
7 |
24,092,777 |
2.00E-05 |
0.058 |
0.03 |
‒6.445 |
|
2_32517 |
7 |
24,094,841 |
2.00E-05 |
0.058 |
0.03 |
6.445 |
|
2_02058 |
7 |
24,097,697 |
2.00E-05 |
0.063 |
0.029 |
6.478 |
|
2_02059 |
7 |
24,097,968 |
2.00E-05 |
0.063 |
0.029 |
‒6.478 |
|
2_41121 |
7 |
24,080,674 |
3.00E-05 |
0.059 |
0.029 |
‒6.388 |
|
2_11217 |
9 |
643,886 |
3.00E-06 |
0.01 |
0.036 |
‒9.86 |
|
Average |
2_06981 |
2 |
21,120,081 |
2.26E-07 |
0.021 |
0.044 |
8.459 |
Table 3Details of SNP loci associated with flowering time in cowpea identified by genome-wide association study using the elastic-net model with selection probability.
Table 3
|
Year |
SNP marker |
Chr. |
Position (bp) |
Selection Probability |
|
2018 |
2_43560 |
1 |
40,154,785 |
0.81 |
|
2_06981 |
2 |
21,120,081 |
0.77 |
|
2_23118 |
3 |
48,325,012 |
0.94 |
|
2_29320 |
3 |
48,359,209 |
0.72 |
|
2_54082 |
8 |
37,954,268 |
0.85 |
|
2019 |
2_54398 |
1 |
24,021,527 |
0.92 |
|
2_07050 |
1 |
23,028,728 |
0.83 |
|
2_00095 |
4 |
1,936,933 |
0.8 |
|
2_28648 |
5 |
2,087,668 |
0.91 |
|
2_23797 |
5 |
2,092,505 |
0.89 |
|
2_06184 |
5 |
2,070,082 |
0.87 |
|
2_54082 |
8 |
37,954,268 |
0.94 |
|
2_12611 |
8 |
37,914,805 |
0.79 |
|
Average |
2_07050 |
1 |
23,028,728 |
0.86 |
|
2_06981 |
2 |
21,120,081 |
0.88 |
|
2_23118 |
3 |
48,325,012 |
0.98 |
|
2_29320 |
3 |
48,359,209 |
0.93 |
|
2_32723 |
3 |
49,330,360 |
0.87 |
|
2_06184 |
5 |
2,070,082 |
0.96 |
|
2_54082 |
8 |
37,954,268 |
0.97 |
|
2_24356 |
8 |
28,163,712 |
0.83 |
|
2_12611 |
8 |
37,914,805 |
0.81 |
|
2_20601 |
9 |
40,855,711 |
0.89 |
Table 4Candidate genes associated with the flowering time in cowpea.
Table 4
|
Chr. |
SNP |
Gene |
Location (bp) |
Gene description |
Flowering function |
Reference |
|
1 |
2_54398 |
Vigun01g084000
|
23841489..23844362 |
Protein FLOWERING LOCUS T/Phosphatidylethanolamine-binding proteinz)
|
Regulation of flowering time |
, Wickland and Hanzawa 2015
|
|
|
Vigun01g084400
|
23914471..23916601 |
PPR repeat |
Expression in flowering |
, Saha et al. 2007
|
|
|
Vigun01g085000
|
24039903..24041896 |
No apical meristem (NAM) protein |
Control of flowering time |
, Nuruzzaman et al. 2013
|
|
|
Vigun01g085500
|
24123050..24132785 |
RNA recognition motif. (a.k.a. RRM, RBD, or RNP domain) |
Regulation of flowering time |
, Lorković and Barta 2002
|
|
2_43560 |
Vigun01g227200
|
39998036..39998455 |
PROTEIN EARLY FLOWERING 4z)
|
Early flowering |
, Khanna et al. 2003
|
|
|
Vigun01g229200
|
40176676..40177200 |
Ring finger domain |
Regulation of flowering time |
, Du et al. 2016
|
|
|
Vigun01g229900
|
40232831..40236901 |
Leucine rich repeat N-terminal domain/Protein tyrosine kinase |
Expression in flowering |
, Forsthoefel et al. 2005
|
|
|
Vigun01g230200
|
40253121..40256700 |
METHYLTRANSFERASE |
Regulation of flowering time |
, Niu et al. 2007
|
|
2 |
2_06981 |
Vigun02g062600
|
20965948..20970544 |
F-BOX/KELCH-REPEAT PROTEIN SKIP11z)
|
Established in circadian clock and flowering time regulation |
, ul Hassan et al. 2015
|
|
|
Vigun02g063000
|
21055222..21057708 |
Homeobox associated leucine zipper |
Regulation of flowering time |
, Tang et al. 2019
|
|
|
Vigun02g063300
|
21109190..21114863 |
Myb-like DNA-binding domain |
Expression in flowering |
, Ambawat et al. 2013
|
|
|
Vigun02g064000
|
21245076..21247100 |
PPR repeat |
Expression in flowering |
, Saha et al. 2007
|
|
|
Vigun02g064100
|
21250319..21252469 |
Ubiquitin-conjugating enzyme |
Regulation of flowering time |
, Wang et al. 2016
|
|
3 |
2_23118 |
Vigun03g295800
|
48232939..48235720 |
Zinc finger, C3HC4 type (RING finger)/Predicted E3 ubiquitin ligase |
Regulation of flowering time |
, Yang et al. 2015
|
|
|
Vigun03g296800
|
48322318..48325239 |
Casein kinase II regulatory subunitz)
|
Regulation of flowering time |
, Ogiso et al. 2010
|
|
5 |
2_06184 |
Vigun05g024100
|
1985451..1988773 |
PPR repeat |
Expression in flowering |
, Saha et al. 2007
|
|
|
Vigun05g024300
|
2001441..2005611 |
Protein kinase domain/Leucine rich repeat N-terminal domain |
Expression in flowering |
, Forsthoefel et al. 2005) |
|
8 |
2_54082 |
Vigun08g218000
|
37770920..37773229 |
RNA recognition motif. (a.k.a. RRM, RBD, or RNP domain) |
Regulation of flowering time |
, Lorković and Barta 2002
|
|
|
Vigun08g220700
|
37932014..37935579 |
PPR repeat |
Expression in flowering |
, Saha et al. 2007
|
|
|
Vigun08g220900
|
37944978..37948715 |
Zinc finger, C3HC4 type (RING finger)/E3 UBIQUITIN-PROTEIN LIGASE UHRF-RELATED |
Regulation of flowering time |
, Yang et al. 2015
|
|
|
Vigun08g222400
|
38033233..38033754 |
VQ motif |
Delayed flowering time |
, Jing and Lin 2015
|
|
|
Vigun08g223500
|
38107101..38107802 |
C2H2-type zinc finger |
Flower development |
, Gourcilleau et al. 2011
|
References
- Alqudah AM, Sallam A, Baenziger PS, Börner A. 2020. GWAS: Fast-forwarding gene identification and cha-racterization in temperate Cereals: lessons from Barley-A review. J. Adv. Res.. 22: 119-135.
- Amasino RM, Michaels SD. 2010. The timing of flowering. Plant Physiol.. 154: 516-520.
- Ambawat S, Sharma P, Yadav NR, Yadav RC. 2013. MYB transcription factor genes as regulators for plant responses: an overview. Physiol. Mol. Biol. Plants. 19: 307-321.
- Andargie M, Pasquet RS, Muluvi GM, Timko MP. 2013. Quantitative trait loci analysis of flowering time related traits identified in recombinant inbred lines of cowpea (Vigna unguiculata). Genome. 56: 289-294.
- Carvalho M, Lino-Neto T, Rosa E, Carnide V. 2017. Cowpea: a legume crop for a challenging environment. J. Sci. Food Agric.. 97: 4273-4284.
- Cho S, Kim K, Kim YJ, Lee JK, Cho YS, Lee JY, et al. 2010. Joint identification of multiple genetic variants via elastic-net variable selection in a genome-wide association analysis. Ann. Hum. Genet.. 74: 416-428.
- Cho S, Kim H, Oh S, Kim K, Park T. 2009. Elastic-net regularization approaches for genome-wide association studies of rheumatoid arthritis. BMC Proc.. 3: 1-6.
- Coulibaly S, Pasquet RS, Papa R, Gepts P. 2002. AFLP analysis of the phenetic organization and genetic diversity of Vigna unguiculata L. Walp. reveals extensive gene flow between wild and domesticated types. Theor. Appl. Genet.. 104: 358-366.
- Du Y, He W, Deng C, Chen X, Gou L, Zhu F, et al. 2016. Flowering-related RING protein 1 (FRRP1) regulates flowering time and yield potential by affecting histone H2B monoubiquitination in rice (Oryza sativa). PLoS One. 11: e0150458.
- Ehlers J, Hall A. 1997. Cowpea (Vigna unguiculata L. Walp.). Field Crops Res.. 53: 187-204.
- Elowad HO, Hall AE. 1987. Influences of early and late nitrogen fertilization on yield and nitrogen fixation of cowpea under well-watered and dry field conditions. Field Crops Res.. 15: 229-244.
- Flint-Garcia SA, Thornsberry JM, Buckler IV ES. 2003. Structure of linkage disequilibrium in plants. Ann. Rev. Plant Biol.. 54: 357-374.
- Forsthoefel NR, Cutler K, Port MD, Yamamoto T, Vernon DM. 2005. PIRLs: A novel class of plant intracellular leucine-rich repeat proteins. Plant Cell Physiol.. 46: 913-922.
- Franklin KA, Quail PH. 2010. Phytochrome functions in Arabidopsis development. J. Exp. Bot.. 61: 11-24.
- Garcia M, Eckermann P, Haefele S, Satija S, Sznajder B, Timmins A, et al. 2019. Genome-wide association mapping of grain yield in a diverse collection of spring wheat (Triticum aestivum L.) evaluated in southern Australia. PLoS One. 14: e0211730.
- Gaut BS, Long AD. 2003. The lowdown on linkage disequilibrium. Plant Cell. 15: 1502-1506.
- Gourcilleau D, Lenne C, Armenise C, Moulia B, Julien J-L, Bronner G, et al. 2011. Phylogenetic study of plant Q-type C2H2 zinc finger proteins and expression analysis of poplar genes in response to osmotic, cold and mech-anical stresses. DNA Res.. 18: 77-92.
- Gupta PK, Rustgi S, Kulwal PL. 2005. Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol. Biol.. 57: 461-485.
- Hiersche M, Rühle F, Stoll M. 2013. Postgwas: advanced GWAS interpretation in R. PLoS One. 8: e71775.
- Ishiyaku MF, Singh BB, Craufurd PQ. 2005. Inheritance of time to flowering in cowpea (Vigna unguiculata (L.) Walp.). Euphytica. 142: 291-300.
- Jaiswal V, Gupta S, Gahlaut V, Muthamilarasan M, Bandyopadhyay T, Ramchiary N, et al. 2019. Genome-wide association study of major agronomic traits in foxtail millet (Setaria italica L.) using ddRAD sequencing. Sci. Rep.. 9: 1-11.
- Jing Y, Lin R. 2015. The VQ motif-containing protein family of plant-specific transcriptional regulators. Plant Physiol.. 169: 371-378.
- Khanna R, Kikis EA, Quail PH. 2003. EARLY FLOWERING 4 functions in phytochrome B-regulated seedling de-etiolation. Plant Physiol.. 133: 1530-1538.
- Keim P, Olson TC, Shoemaker RC. 1988. A rapid protocol for isolating soybean DNA. Soybean Genet. Newsl.. 15: 150-152.
- Kim K, Koo J, Sun H. 2020. An empirical threshold of selection probability for analysis of high-dimensional correlated data. J. Stat. Comput.. 90: 1606-1617.
- Kim MY, Shin JH, Kang YJ, Shim SR, Lee S-H. 2012. Divergence of flowering genes in soybean. J. Biosci.. 37: 857-870.
- Kongjaimun A, Kaga A, Tomooka N, Somta P, Vaughan DA, Srinives P. 2012. The genetics of domestication of yardlong bean, Vigna unguiculata (L.) Walp. ssp. unguiculata cv.-gr. sesquipedalis. Ann. Bot.. 109: 1185-1200.
- Li M, Li C, Guan W. 2008. Evaluation of coverage variation of SNP chips for genome-wide association studies. Eur. J. Hum. Genet.. 16: 635-643.
- Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, et al. 2012. GAPIT: genome association and prediction inte-grated tool. Bioinformatics. 28: 2397-2399.
- Liu YY, Yang KZ, Wei XX, Wang XQ. 2016. Revisiting the phosphatidylethanolamine-binding protein (PEBP) gene family reveals cryptic FLOWERING LOCUS T gene homologs in gymnosperms and sheds new light on functional evolution. New Phytol.. 212: 730-744.
- Lo S, Muñoz-Amatriaín M, Boukar O, Herniter I, Cisse N, Guo Y-N, et al. 2018. Identification of QTL controlling domestication-related traits in cowpea (Vigna unguiculata L. Walp). Sci. Rep.. 8: 1-9.
- Lonardi S, Muñoz-Amatriaín M, Liang Q, Shu S, Wanamaker SI, Lo S, et al. 2019. The genome of cowpea (Vigna unguiculata [L.] Walp.). Plant J.. 98: 767-782.
- Lorković ZJ, Barta A. 2002. Genome analysis: RNA re-cognition motif (RRM) and K homology (KH) domain RNA-binding proteins from the flowering plant Arabidopsis thaliana. Nucl. Acids Res.. 30: 623-635.
- Mammadov J, Aggarwal R, Buyyarapu R, Kumpatla S. 2012. SNP markers and their impact on plant breeding. Int. J. Plant Genomics. 2012: 728398.
- Meinshausen N, Bühlmann P. 2010. Stability selection. J. R. Stat. Soc. Series B Stat. Methodol.. 72: 417-473.
- Niu L, Lu F, Pei Y, Liu C, Cao X. 2007. Regulation of flowering time by the protein arginine methyltransferase AtPRMT10. EMBO Rep.. 8: 1190-1195.
- Nuruzzaman M, Sharoni AM, Kikuchi S. 2013. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol.. 4: 248
- Ogiso E, Takahashi Y, Sasaki T, Yano M, Izawa T. 2010. The role of casein kinase II in flowering time regulation has diversified during evolution. Plant Physiol.. 152: 808-820.
- Perrino P, Laghetti G, Zeuli PS, Monti L. 1993. Diversification of cowpea in the Mediterranean and other centres of cultivation. Genet. Resour. Crop Evol.. 40: 121-132.
- Roberts E, Summerfield R, Ellis R, Qi A. 1993. Adaptation of flowering in crops to climate. Outlook Agric.. 22: 105-110.
- Saha D, Prasad A, Srinivasan R. 2007. Pentatricopeptide repeat proteins and their emerging roles in plants. Plant Physiology and Biochemistry. 45: 521-534.
- Schulz-Streeck T, Ogutu JO, Karaman Z, Knaak C, Piepho HP. 2012. Genomic selection using multiple populations. Crop Sci.. 52: 2453-2461.
- Seo E, Kim K, Jun TH, Choi J, Kim SH, Muñoz-Amatriaín M, et al. 2020. Population Structure and Genetic Diversity in Korean Cowpea Germplasm Based on SNP Markers. Plants. 9: 1190
- Song YH, Shim JS, Kinmonth-Schultz HA, Imaizumi T. 2015. Photoperiodic flowering: time measurement mech-anisms in leaves. Ann. Rev. Plant Biol.. 66: 441-464.
- Tang Y, Liu X, Wang J, Li M, Wang Q, Tian F, et al. 2016. GAPIT version 2: an enhanced integrated tool for genomic association and prediction. Plant Genome. 9: 1-9.
- ul Hassan MN, Zainal Z, Ismail I. 2015. Plant kelch containing F-box proteins: structure, evolution and functions. RSC Adv.. 5: 42808-42814.
- Uzun S. 2006. The quantitative effects of temperature and light on the number of leaves preceding the first fruiting inflorescence on the stem of tomato (Lycopersicon esculentum, Mill.) and aubergine (Solanum melongena L.). Sci. Hortic.. 109: 142-146.
- VanRaden PM. 2008. Efficient methods to compute genomic predictions. J. Dairy Sci.. 91: 4414-4423.
- Varshney RK, Nayak SN, May GD, Jackson SA. 2009. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol.. 27: 522-530.
- Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 2017. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet.. 101: 5-22.
- Waldmann P, Mészáros G, Gredler B, Fuerst C, Sölkner J. 2013. Evaluation of the lasso and the elastic net in genome-wide association studies. Front. Genet.. 4: 270
- Wang S, Cao L, Wang H. 2016. Arabidopsis ubiquitin-conjugating enzyme UBC22 is required for female gametophyte development and likely involved in Lys11-linked ubiquitination. J. Exp. Bot.. 67: 3277-3288.
- Wickland DP, Hanzawa Y. 2015. The FLOWERING LOCUS T/TERMINAL FLOWER 1 gene family: functional evolution and molecular mechanisms. Mol. Plant. 8: 983-997.
- Wimmer V, Albrecht T, Auinger H, Schon C. 2012. Synbreed: a framework for the analysis of genomic prediction data using R. Bioinformatics. 28: 2086-2087.
- Wimmer V, Lehermeier C, Albrecht T, Auinger H-J, Wang Y, Schön C-C. 2013. Genome-wide prediction of traits with different genetic architecture through efficient variable selection. Genetics. 195: 573-587.
- Xu G, Ma H, Nei M, Kong H. 2009. Evolution of F-box genes in plants: different modes of sequence divergence and their relationships with functional diversification. Proc. Natl. Acad. Sci. U. S. A.. 106: 835-840.
- Xu P, Wu X, Wang B, Hu T, Lu Z, Liu Y, et al. 2013. QTL mapping and epistatic interaction analysis in asparagus bean for several characterized and novel horticulturally important traits. BMC Genet.. 14: 4
- Xu P, Wu X, Wang B, Luo J, Liu Y, Ehlers J, et al. 2012. Genome wide linkage disequilibrium in Chinese asparagus bean (Vigna. unguiculata ssp. sesquipedialis) germplasm: implications for domestication history and genome wide association studies. Heredity. 109: 34-40.
- Xu Z, Taylor JA. 2009. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucl. Acids Res.. 37: W600-W605.
- Yang N, Lu Y, Yang X, Huang J, Zhou Y, Ali F, et al. 2014. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet.. 10: e1004573.
- Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, et al. 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet.. 38: 203-208.
- Zhang Z, Ersoz E, Lai C-Q, Todhunter RJ, Tiwari HK, Gore MA, et al. 2010. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet.. 42: 355-360.
- Zhao X, Teng W, Li Y, Liu D, Cao G, Li D, et al. 2017. Loci and candidate genes conferring resistance to soybean cyst nematode HG type 2.5.7. BMC Genomics. 18: 1-10.
- Zou H, Hastie T. 2005. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Series B Stat. Methodol.. 67: 301-320.