Abstract
Functional stay-green (FSG) delays leaf yellowing, maintaining photosynthetic competence, whereas nonfunctional stay-green (NFSG) retains only leaf greenness without sustaining photosynthetic activity. Retention of chlorophylls and photosynthetic capacity is important for increasing crop yield. We determined the main-effect quantitative trait loci (QTLs) for FSG traits in the japonica rice SNU-SG1 and isolated candidate genes. To identify QTLs influencing FSG, we analyzed eight traits: (1) 1 day after heading-degree of chlorophyll content of flag leaf, (2) 1 day after heading-degree of chlorophyll content of second leaf, (3) 1 day after heading-degree of chlorophyll content of flag and second leaves, (4) 50 day after heading-degree of chlorophyll content of flag leaf, (5) 50 day after heading-degree of chlorophyll content of second leaf, (6) 50 day after heading-degree of chlorophyll content of flag and second leaves, (7) relative decline degree of chlorophyll content of flag and second leaves, and (8) flowering time. We carried out QTL analysis with F7 RIL from a cross of japonica rice ‘SNU-SG1’ and indica rice ‘Milyang23 (M23)’. Using 131 molecular markers, we identified 18 QTLs for the eight traits with a threshold LOD value > 2.8. Sequence analysis identified 16 candidate genes for 10 main-effect QTLs. Of these, we have chosen seven strong candidate genes for the 10 main-effect QTLs. These genetic resources will be useful for breeding high-yielding rice cultivars.
-
Key words: Candidate gene, Functional stay-green, Heading date, Quantitative trait locus, Rice, Whole genome re-sequencing
INTRODUCTION
Rice (
Oryza sativa L.) is one of the most essential cereal crops in Asia. Increasing yield of cereals has been among the most important goals of crop breeding and plant science researches. To increase crop yield, plants require not only directly linked high yield potential, which is defined as yield components (grain weight, panicle number per plant, and grain number per panicle), but also an indirectly linked yield potential traits that, to a large extent, is determined by photosynthetic efficiency and flowering time. Photosynthesis after heading accounts for 60–90% of the total carbon accumulated in panicles at harvest, while around 80% of total panicle nitrogen uptake happens before heading and is continuously remobilized from leaves to grains during senescence (
Mae 1997; Yue
et al. 2006).
Delayed senescence or stay-greenness during the last stage of plant development has been thought an important trait for increasing crop production by source strength, and its genetic and physiological bases have been studied in a number of plant species. Stay-green phenotypes are classified into five distinguishable types, A to E, according to their senescing types (
Thomas and Howarth 2000). Stay-greenness also can be divided typically into two groups, nonfunctional and functional. FSG is defined as keeping both leaf greenness and photosynthetic ability much longer during post-flowering stage while NFSG keeps only leaf greenness without photosynthetic competence.
Park and Lee (2003) introduced a stay-green variant from the rice collections, ‘SNU-SG1’, which is classified as the type B FSG rice which starts leaf senescence normally but thereafter photosynthetic capacity and chlorophyll content decrease slower than the other elite rice cultivars after heading.
Stay-green trait has been studied in many crop plants, such as maize (
Gentinetta et al. 1986), durum wheat (
Spano et al. 2003), sorghum (
Walulu 1994), soybean (
Pierce et al. 1984), and potato (
Schittenhelm et al. 2004). FSG genotypes in durum wheat were reported to maintain higher activity of photosynthesis and higher grain weights than the parental genotype (
Spano et al. 2003). FSG trait of SNU-SG1 contributed to increasing grain yield through enhancing seed-setting rates. The SNU-SG1 stay-green trait was positively correlated with grain yield and seed-setting rate, but has no significant correlation with other yield components including tillers per plant, grains per panicle, or grain weight (
Yoo et al. 2007).
Flowering time or heading date is a determining factor enabling crops to adapt to seasonal changes, especially in the northernmost areas of rice cultivation, and for photoperiod sensitivity. Signaling mechanisms for repression of flowering or photoperiod-dependent promotion revealed rice has both conserved unique Ehd1 and Hd1 pathways to control the floral integrators,
Hd1 and
Hd3a expression (
Yano et al. 2000;
Kosima et al. 2002), which act as rice florigen orthologs of
CO and
FT in
Arabidopsis thaliana, respectively.
Candidate gene cloning and QTL mapping using DNA polymorphic markers are useful tools to understand the genetic basis of complicated phenotypic variation; moreover, the application of molecular markers developed for QTL analysis improves breeding efficiency by enabling marker-assisted selection (MAS) for variable agronomic traits. However, traditional approaches for QTL analysis are labor-intensive and time-consuming because of the low (>15%) efficiency of polymorphic markers (
Lim et al. 2014) and the difficulties in the determination of candidate genes. With the advent of next-generation sequencing technology (NGS), these limitations can be overcome by applying whole genome sequencing (WGS) technique (
Lim et al. 2014). Recently, another new approach named Genotyping-by-Sequencing (GBS), a simple highly-multiplexed system, is designed for rapid QTL identification for a given phenotype by generating large numbers of SNPs for use in the genetic analyses.
In this study, we conducted QTL mapping for the seven traits related to FSG in rice and identified ten main- and eight minor-effect QTLs using NGS technology. Through resequencing data, we identified seven strong candidate genes for the 10 main-effect QTLs. The FSG and heading date (HD) QTL and the candidate genes information would be useful for breeding for high yielding rice cultivars.
MATERIALS AND METHODS
Mapping population and growth conditions
To identify QTL influencing degree of chlorophyll content after heading in rice, a high-yielding
indica/japonica hybrid cultivar M23 was crossed with a
japonica cultivar FSG rice SNU-SG1 (
Yoo et al. 2007). Among 338 F
7 RILs developed from the F
2 population by single seed descent (SSD), 178 were chosen for subsequent QTL mapping; these include 40 lines showing extreme phenotypic values for each trait. Field experiment was managed following the standard rice cultivation practices in Suwon, South Korea (37° N latitude). Seeds were sown on April 20 and seedlings were transplanted on May 19 at spacing of 0.3 m by 0.15 m with one seedling per hill in 2010 and 2011. Compound fertilizer (21N-17P-17K) was applied in the same amount (24 kg/10a) and applied three times: before transplanting, and at seven and ten weeks after transplanting.
For the phenotypic evaluation of the selected 178 F7 RILs, we measured the agronomic traits. The chlorophyll content of the flag and second leaves of each F7 line were measured between 12:00 and 15:00, at heading and 50 days after heading, with a Minolta Chlorophyll Meter SPAD-502 (Minolta, Japan), an indirect indicator of chlorophyll content. To ensure measurements were taken from the right tiller and day, tillers were tagged at the day of heading. SPAD reading was performed with three replications by measuring three panicles per plant and at least three parts of the leaf for all of the stay-green traits. The degree of chlorophyll content (SPAD value) of the flag and second leaves measured at the day after heading were designated as degree of chlorophyll content of flag leaf (DCF) and degree of chlorophyll content of flag leaf (DCS), respectively. DCFS was derived from the average of DCF and DCS to complement the FSG trait. Relative decline degree of chlorophyll content (RDCFS) in the flag and second leaves was shown using the SPAD value of 50 days after flowering (S50) divided by SPAD value of one day after flowering (S1), designated RDCF, RDCS and RCFS, respectively. RDCFS was calculated as (S50 / S1) × 100 %. HD were recorded from sowing to the appearance of the first panicle of about 1 cm in length.
Molecular markers for genotyping
Genomic DNAs were extracted from leaf tissues by a CTAB method (Murray and Thompson 1980). Of 131 DNA markers used for genotyping, 68 are RM-series that were designed by Temnykh et al. (2001), and 35 markers are S-series that were designed based on the sequence differences between japonica and indica, using information from the Crop Molecular Breeding Lab, Seoul National University (unpublished). The other 28 markers are NGS-based insertion/deletion (NID) markers designed by sequence comparison of insertions/deletions (InDels) between the two parent genotypes. The size difference of the NID markers was around 7~11 bp.
Linkage map construction and data analysis
A molecular linkage map was constructed using Mapmaker 3.0 (Lander et al. 1987; Lincoln et al. 1993). The distance between markers is presented in centiMorgans (cM) using the Kosambi map function (Kosambi 1944), and order of markers was established using three point linkage analyses. QTL analysis was conducted with the composite interval mapping (CIM) implemented in Qgene 4.3.10 software (Joehanes and Nelson 2008). Significance thresholds for QTL detection were determined with 1000 permutations, and the LOD threshold for each QTL ranged from 2.24 to 3.03 with 95% confidence. The QTLs explaining more than 10% of the phenotypic effect were defined as main-effect QTLs.
High-throughput whole genome re-sequencing (WGS)
High-throughput WGS was performed on the two parents, SNU-SG1 and M23. The Illumina Genome Analyzer II was used to generate short reads, and bases were called with the Sequence Analysis Software of Pipeline version 1.4 of the Genome Analyzer (Illumina, Inc. San Diego, CA). Genomic DNAs were extracted using the QIAquick PCR Purification Kit (Qiagen) from leaf tissues. Full sequencing was processed at the National Instrumentation Center for Environmental Management (NICEM) at Seoul National University, Seoul, Korea. We ultimately gained 26X and 31X fold coverage as well as 9 Gbp and 11 Gbp in read length for SNU-SG1 and M23, respectively. Then, the 75 bp paired-end reads of the two parents were mapped to the japonica cv. Nipponbare reference genome. Finally, the low-quality bases (Q score in scale < 20), sites with conflicting genotypes among reads were excluded, and only the reads aligned to unique locations in the reference genome were used for sequence construction. The total number of SNPs was 3,202,922, which include 163,933 from SNU-SG1 and 3,038,989 from M23.
Identification of candidate genes in QTL regions using WGS data
Of the 18 QTLs, 10 main-effect QTLs with high LOD and R
2 value were used for candidate gene analysis. We identified candidate genes containing mutated sequences such as SNPs and InDels by the following process. First, a reference sequence (Nipponbare) corresponding to each main-effect QTL was downloaded from the GRAMENE database (
http://www.gramene.org/). The genomic research regions associated with each QTLs were selected by confidence interval that was determined by 1.5-LOD support interval. An additional 0.5 Mb sequences from the two flanking DNA markers covering the target QTL were analyzed to increase accuracy. Coding sequence regions derived from SNU-SG1 and M23 were interrogated to detect mutated sequences using the AutoHotkey macro program by work sheet (
Lim et al. 2014). Among the genes harboring mutated sequences, only the genes for which the mutation caused amino acid changes were selected as candidate genes.
RESULTS
Phenotypic variation in F7 RILs from the M23/SNU-SG1 cross
For QTL analysis of the eight traits related to FSG and flowering time in rice, we used F
7 178 RILs from the cross of SNU-SG1 (
japonica) and M23 (
indica) (
Lim et al. 2014). Phenotypic variation was evaluated in the SNU-SG1, M23 and the RILs (
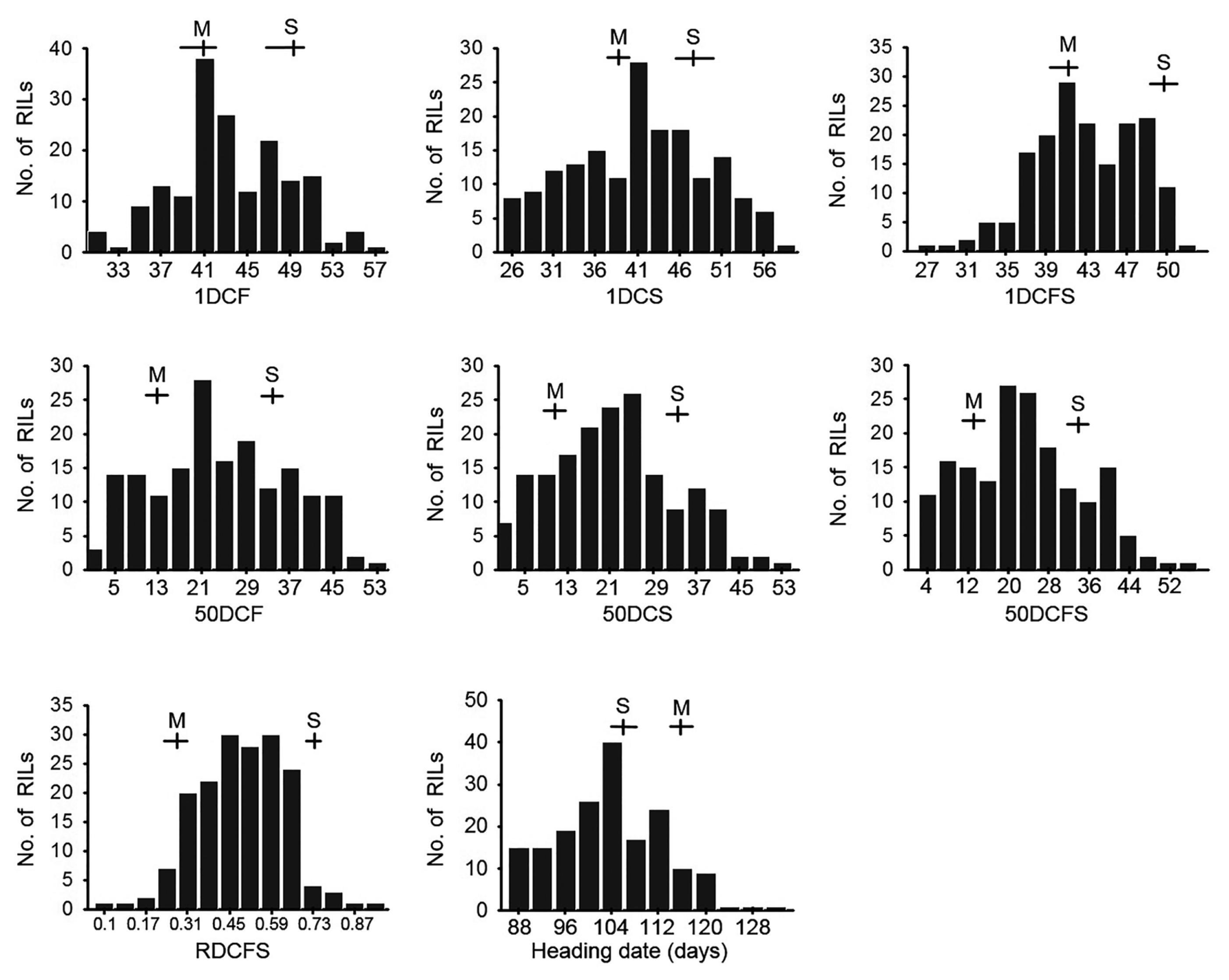
Fig. 1). Most of the traits showed bidirectional transgressive segregation with approximately normal distribution. A large amount of variation was observed between the two parental cultivars for most of the traits, except for RDCFS. Descriptive statistics of the eight traits is shown in
Table 1. The correlation among the traits was analyzed in the F
7 RIL population (
Table 2). A positive correlation was observed between RDCFS and other traits except for HD. Relatively high positive correlations were observed between RDCS, 50 days after chlorophyll content related traits (50DCF, 50DCS and 50DCFS), and more than one day after chlorophyll content associated traits (1DCF, 1DCS and 1DCFS). A negative correlation was observed between HD and other seven FSG traits.
A coarse-scale linkage map was originally constructed over the whole rice genome using F
7 178 RILs, and 35 STS and 68 SSR markers. To construct linkage map for the separated regions in the original map, we designed 28 whole genome re-sequencing data-based InDel markers. We used Mapmaker 3.0 to construct the genetic linkage map, which consisted of 10 linkage groups and covered 2,451 cM. Each adjacent marker’s average distance was 14.5-centimorgan (cM), which is less than 20 cM, the minimal necessary level for QTL analysis (
Lander and Botstein 1989).
QTLs were identified based on the phenotypic effect (R
2) and LOD threshold value (see Materials and Methods). As a result, a total of 18 QTLs for the eight traits, with 6.9 to 20.4% phenotypic effect (R
2) and 2.8 to 8.83 LOD values, across all the chromosomes except chromosomes 1, 2, 4, 7 and 12 (
Table 3) were identified. A QTL with highest LOD (8.83) and phenotypic effect (20.4%) was detected for HD on the short arm of chromosome 6 (
Fig. 2,
Table 3). Overall, we detected 2 to 3 QTLs for each functional stay green and flowering time trait. Of the 18 QTLs, 10 accounted for more than 10% of the phenotypic effect and were considered main-effect QTLs, which include
1Dcf5 and
1Dcf9 for 1DCF,
1Dcs5 and
1Dcs9 for 1DCS,
1Dcfs5 and
1Dcfs9 for 1DCFS,
50Dcf9 for 50DCF,
50Dcs9 for 50DCS, and
50Dcfs9 for 50DCFS (
Table 3). Especially, three QTLs (
1Dcs5, 1Dcfs5 and
Hd6) accounted for more than 15% of the phenotypic effect, implying that these genetic materials are valuable resource for the identification of QTL genes affecting FSG and flowering time.
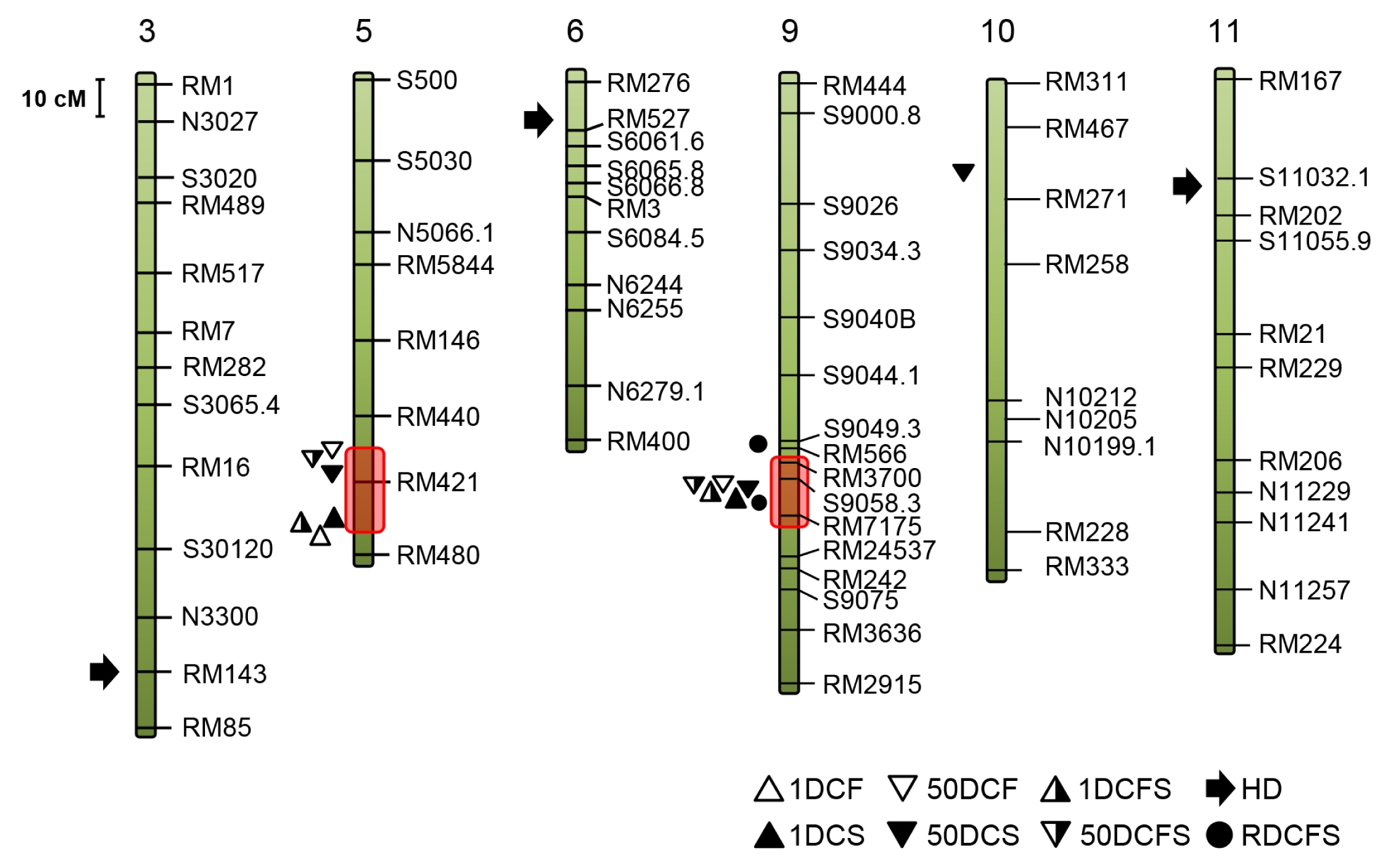
Two QTL hotspots in the genome for multiple traits (
Neto et al. 2012), were identified on chromosomes 5 and 9 (red blocks in
Fig. 2). One, approximately 8 cM window on the long arm of chromosome 9, include six main-effect QTLs (
1Dcs9, 1Dcfs9, 50Dcf9, 50Dcs9,
50Dcfs9 and
Rdcfs for 1DCS, 1DCFS, 50DCF, 50DCS, 50 DCFS and RDCFS, respectively). The other, in a window spanning around 15 cM on chromosome 5, carries four QTLs, including three main-effect QTL (
1Dcs, 50Dcs and
1Dcfs for 1DCS, 50DCS and 1DCFS) and one minor-effect QTL (
50Dcfs for 50DCFS). To compare the physical locations of the QTLs identified in reports and the ones identified in this study, we surveyed the rice research paper for cases in which mapping populations made by the crosses of
japonica and
indica cultivars were used for QTL analysis. We compared the physical locations between the QTLs in the previously published QTLs and in the current study by location of DNA markers. Among the 18 QTLs identified in this study, seven were found to have a matching chromosomal location with those of other reports (
Table 4). The remaining 11 QTLs did not overlap with the published QTLs, which are probably novel QTLs for the traits. Especially, the new 11 QTLs are main-effect QTLs (> 10% phenotypic effect), including
1Dcf5, 1Dcf9, 1Dcs5, 1Dcs9, 1Dcfs9, 50Dcf5, 50Dcf9, 50Dcs5, 50Dcs9, 50Dcfs5, and
50Dcfs9 (
Table 3).
To obtain nucleotide polymorphism data set of cultivar-specific, whole genome resequencing was performed (see Methods) for M23 and SNU-SG1 cultivar. As a reference sequence, a Nipponbare
japonica cultivar, whose whole genome sequence was analyzed by
International Rice Genome Sequencing Program (2005), was appropriated. The polymorphic nucleotide sequences including InDels and substitution were identified by comparing the whole genomic sequences of SNU-SG1 and M23 with the reference sequence (Nipponbare) using Pipeline version 1.4. Out of a total of 363,845 SNPs, 33,527 were identified in SNU-SG1, and 330,317 in M23. Most SNPs in M23 were derived from sequence variation between
japonica and
indica type subspecies. We analyzed all the SNU-SG1 SNPs for candidate gene identification. Especially, the InDels and SNPs capable of changed protein function were considered for M23 to filter out meaningless sequence variation between
indica and
japonica genomes.
For the candidate genes identification, ten main-effect QTLs were selected out of 18 identified QTLs for the eight traits. We analyzed the genomic regions that were selected based on confidence interval for each QTL peak (
Table 3). Confidence intervals (CI) were determined by 1.5 LOD support interval that provides a QTL coverage probability of nearly 95% (
Dupuis et al. 1999). The 95% CI of the QTLs detected in the integrated map using composite interval mapping are given in
Table 3. We determined the specific loci of polymorphic sequences in the main-effect QTL included genes through blast analysis of SNU-SG1 resequence data and reference sequence. Analysis of coding sequence revealed 16 genes harboring 3,161 SNPs in protein coding sequence. Of which, only 165 SNPs induced amino acid mutation (
Table 5).
Among the 16 candidate genes for ten main-effect QTLs, significant candidate genes were segregated by analyzing protein coding sequences change. Among the candidate genes, seven strong candidate genes were identified based on critical SNPs leading to strong mutations, a premature stop codon, frame shift or amino acid changes, in conserved domain sequences (
Table 6). These seven strong candidate genes represented all the traits examined.
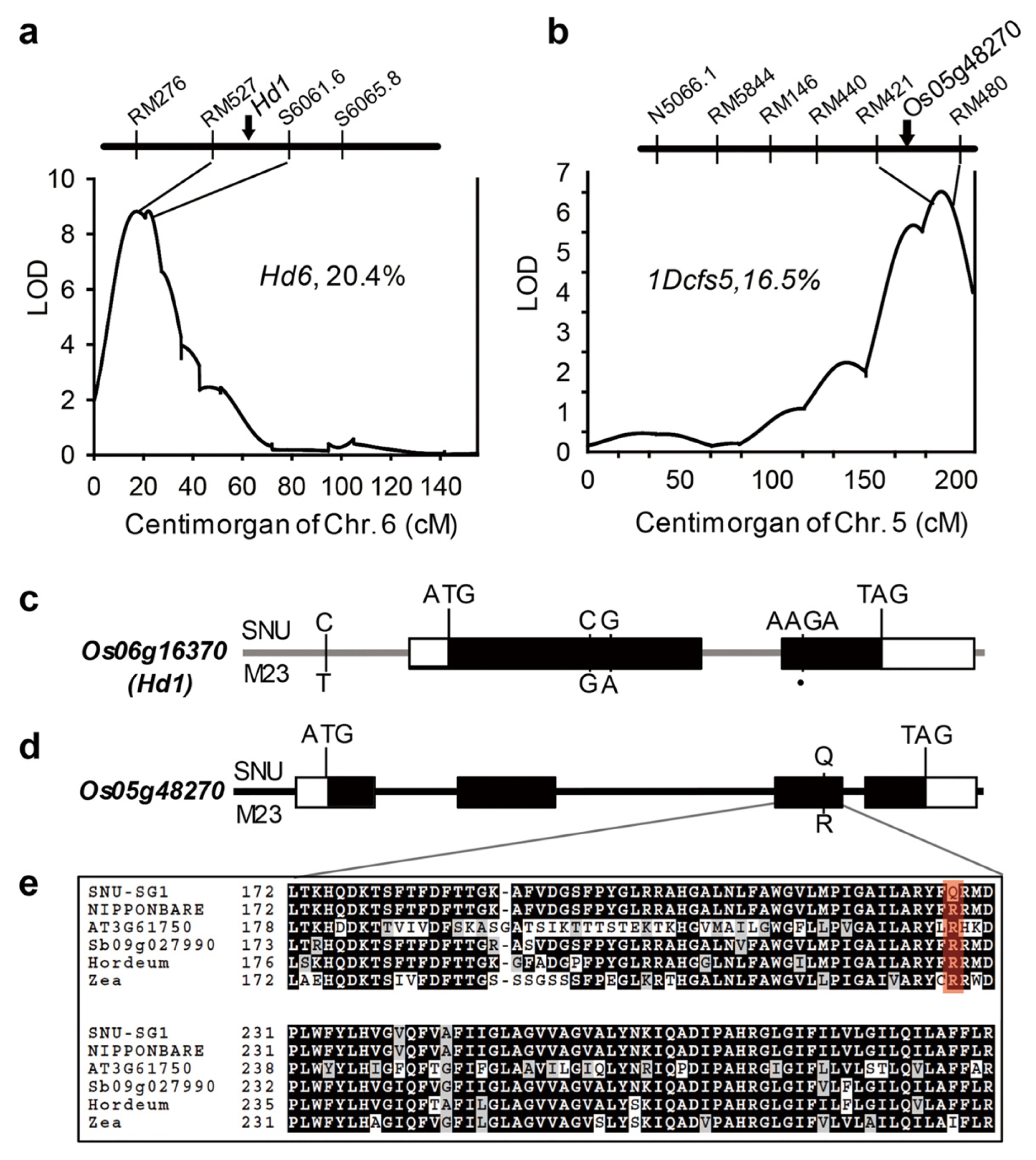
One candidate gene, Os06g16370,
Heading date 1 (
Hd1) was located in the main-effect QTL Hd6 (R
2 = 20.4%) for HD on Chr. 6.
Hd1 controls flowering time by
Hd3a expression regulation in response to photoperiod (
Hayama et al. 2003;
Yano et al. 2000). By resequencing data analysis, we found 4 base-pair nucleic acids deletion in the M23 alleles which creates premature stop codon in protein coding region (
Fig. 4). We further studied the orthologs of these genes in
Arabidopsis thaliana, using TAIR (
http://www.arabidopsis.org/) databases and Gramene (
http://www.gramene.org/). We found three
Arabidopsis orthologs in which three genes have been functionally characterized (
Table 6). One Hd4 QTL, At5g15840, CONSTANS (CO), encodes zinc finger transcription factors, which functions in regulation of flowering under long days. It acts upstream of FT and SOC1 (
Puttenrill et al. 1995). Another 1Dcf, 1Dcs, 1Dcfs, 50Dcf, 50Dcs and 50Dcfs QTL, At1g05300 encodes ZINC TRANSPORTER 5 PRECURSOR (ZIP5), a member of Fe (II) transporter isolog family. This gene acts in zinc II ion transmembrane transport (
Weber et al. 2004). The other 50Dcfs QTLs, At3g53180, the nodulin/glutamine synthetase-like protein (NodGS) is reported to be the key morphogenetic factor (
D’Souza et al. 2001) in vegetative differentiation, playing a role for NodGS in root morphogenesis and microbial elicitation (
Doskocilava et al. 2011).
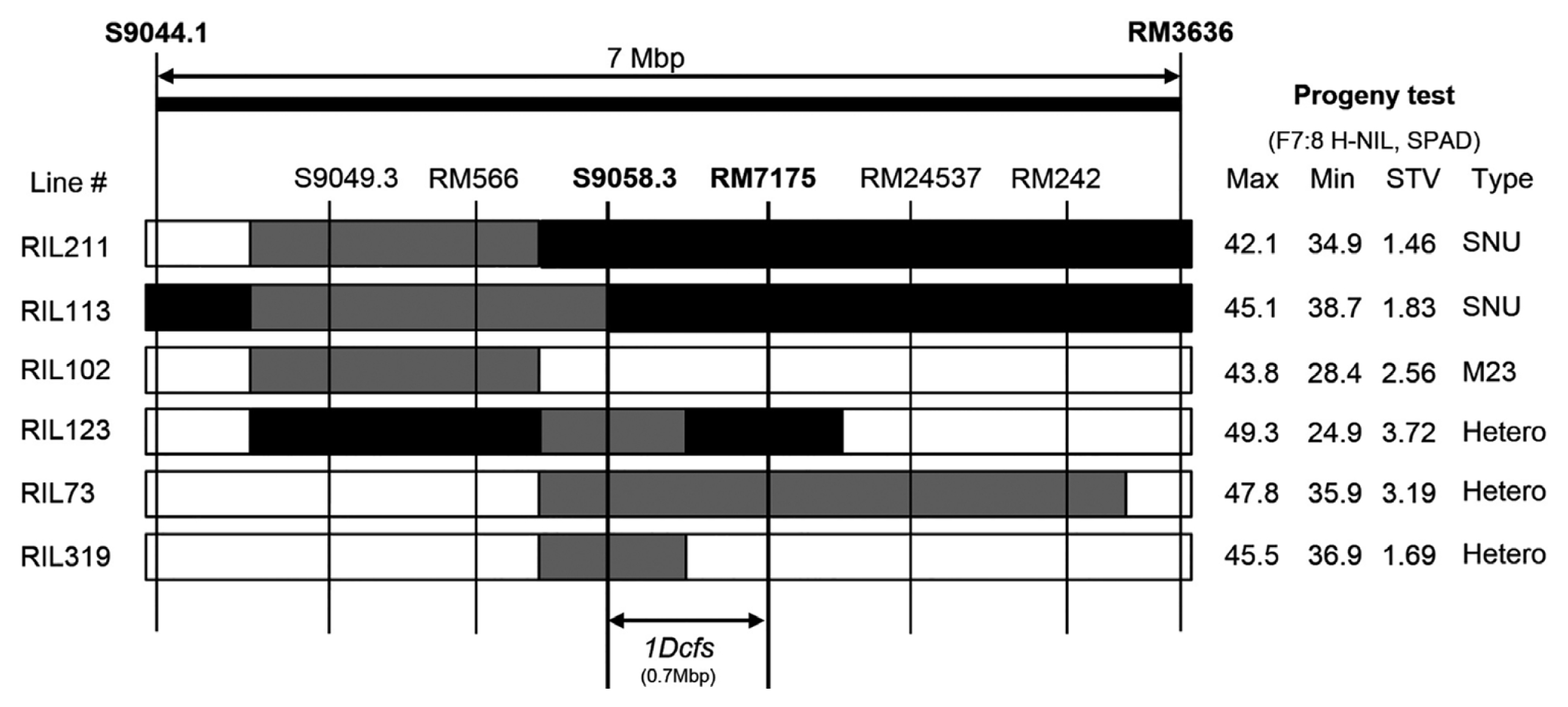
To further validate the QTLs associated with seven significant candidate genes, we developed F
7:8 H-NILs with segregating phenotypes for corresponding traits (
Tuinstra et al. 1997). In the previous study (
Routaboul et al. 2012), phenotypic and genotypic characterization of the H-NIL, progeny of F
7 RIL, allowed identification of the candidate genes underlying main-effect QTLs. In F
7 178 RILs, we selected heterogeneous lines for at least one of the flanking marker loci in the QTL regions (
Fig. 3). Several H-NILs for ten main-effect QTLs harboring strong candidate genes were validated through phenotypic analysis of RILs. Most of the H-NILs, including the F
7 RIL #123-derived ones, showed the highest variation in SPAD value (
Fig. 3). This implicates that target genes are co-located in the corresponding QTL regions, supporting the fidelity of strong candidate genes. Further fine mapping using the identified H-NIL populations may lead to isolation of the correct candidate gene in the main-effect QTLs.
DISCUSSION
A comparison of the previously reported QTLs with identified QTLs in this study
Analyzing candidate genes and their functions from Arabidopsis thaliana orthologs
Resequencing data analysis results in the selection of 16 genes among the 18 identified QTL regions (
Table 5). Seven significant candidate genes were identified through AA sequence analysis for containing SNPs that led to a premature stop codon, frameshift, and amino acid substitution in the conserved domain sequence (
Table 6). An example is the
Hd6 QTL (
Fig. 4A) which is populated with nine genes containing polymorphic SNPs in the protein coding sequences (
Table 5). Of these SNPs, a 4 base-pair nucleic acid deletion in the protein coding region of Os06g16370 (
HD1) occurred in the M23 allele, leading to a frame shift (
Fig. 4C), whereas synonymous SNPs, which do not cause critical mutation, were found for the other eight genes. These results support
HD1 as the likely representative candidate gene for the
Hd6 QTL.
The other QTL,
1Dcfs5, harbors two genes possessing critical mutations in the coding sequence (
Fig. 4B). One of them, Os05g48270, a DOMON-related domain and Cytochrome b561/ferric reductase transmembrane protein coding sequence, contains single SNP which caused critical amino acid substitution (
Fig. 4D) in highly conserved Cytochrome b561 domain (
Fig. 4E). Because this amino-acid substitution is located in the highly conserved domain, it is expected that the function of Os05g48270 protein deteriorated. In the same way, we have identified seven strong candidate genes for the other 10 main-effect QTL (
Table 5).
The putative functions of uncharacterized genes in rice could be inferred from the orthologs in
Arabidopsis thaliana because proteins in these species generally have conserved biochemical and molecular functions. For example, FLAVIN-BINDING, KELCH REPEAT, F-BOX 1 (FKF1) is activated by blue light and promotes flowering through the transcriptional and post-translational regulation of CONSTANS under inductive long day conditions, in
Arabidopsis thaliana.
Oryza sativa L. orthologs gene
OsFKF1 also interacts with OsGI and OsCDF1 and conducts similar and distinct roles of
FKF1 in
Arabidopsis and rice (
Han et al. 2015).
The functions of three
Arabidopsis orthologs have been reported. The
Arabidopsis CONSTANS (CO), nuclear zinc-finger protein, functions as flowering initiators in response to day length pathway, and in response to the transcription of
FT that encodes a protein of RAF-kinase-inhibitor-like (
An et al. 2004). A rice ortholog,
Heading Date 1 (
HD1) encoding a putative CCT/B-box zinc finger protein, has been reported to regulate transcription activation because of zinc finger domain. This indicates that
Hd1 is a promising candidate gene related to the
Hd6 QTL detected for flowering time.
Another candidate gene, Os05g39540, located in the region of
1Dcf5, 1Dcs5, 1Dcfs5, 50Dcf5, 50Dcs5 and
50Dcfs5 (
Table 3), is a rice ortholog of
Arabidopsis ZINC TRANSPORTER 5 PRECURSOR, ZIP5, encoding a Zinc/Iron permease, which is involved in the zinc II ion transmembrane transport. The greater zinc accumulation did not affect the chlorophyll content of
T. caerulescens shoots. But accumulation of lower Zn levels in
T. arvense shoots resulted in dramatic leaf chlorosis as indicated by a significant reduction in RDCFS (Mitch
et al. 1999). If similar functions existed in rice, decreased levels of Zn could affect chlorophyll level during leaf development process. These suggest that this gene is a promising candidate for
1Dcf5, 1Dcs5, 1Dcfs5, 50Dcf5, 50Dcs5 and
50Dcfs5 related FSG traits.
The gene Os10g31820, located in the region of
50Dcf10 QTL, is an ortholog of AT3G53180, which encodes a protein that is the product of a fusion gene with a C-terminal GSI like sequence and an N-terminal part sharing homology with nodulins. Down-regulated NodGS in RNAi plant caused disrupted root cap development and short main root (
Doskocilava et al. 2011). Root development is important for whole plant growth including leaf development. Interruption of root development might eventually affect yield and yield related components. Os10g31820 is a significant candidate gene for
50Dcf10 influencing leaf greenness.
In summary, we conducted QTL analysis for FSG and flowering time, and did candidate gene identification using resequence data of the SNU-SG1 and M23. We detected a total of 18 QTLs for eight traits. The 11 novel QTLs, two QTL hotspots and 7 significant candidate genes identified in this study offer valuable data for breeding for high yield in rice.
ACKNOWLEDGMENTS
This work was carried out with the support of “Next-Generation BioGreen21 Program for Agriculture & Technology Development (Project No. PJ01106301)”, Rural Development Administration, Republic of Korea.
Fig. 1Frequency distribution of the eight traits in the 178 F7 RILs derived from the cross of SNU-SG1/M23. The vertical axis of each figure indicates the number of F7 RILs. Means and ranges of the parents are marked at the top of each histogram; S, SNU-SG1 and M, M23.
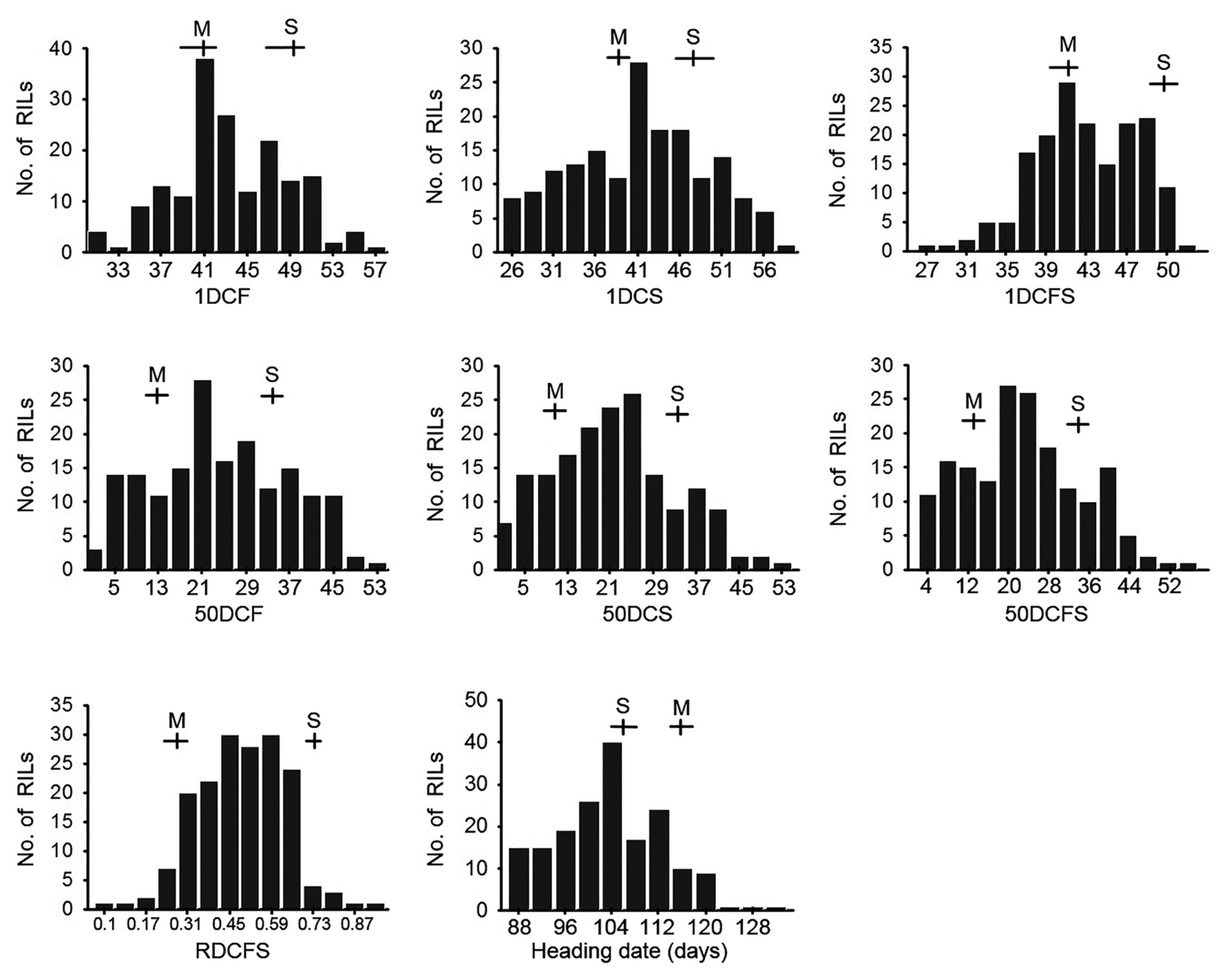
Fig. 2Chromosomal locations of QTLs detected by Q-genes in the F7 RIL population of SNU-SG1/M23. Chromosomes are numbered at the top and markers are listed on the right of each chromosome. Various geometric figures indicate the location of peak LODs of the eight traits. Chromosome 1, 2, 4, 7, 8 and 12 are not shown because QTL were not detected on them.
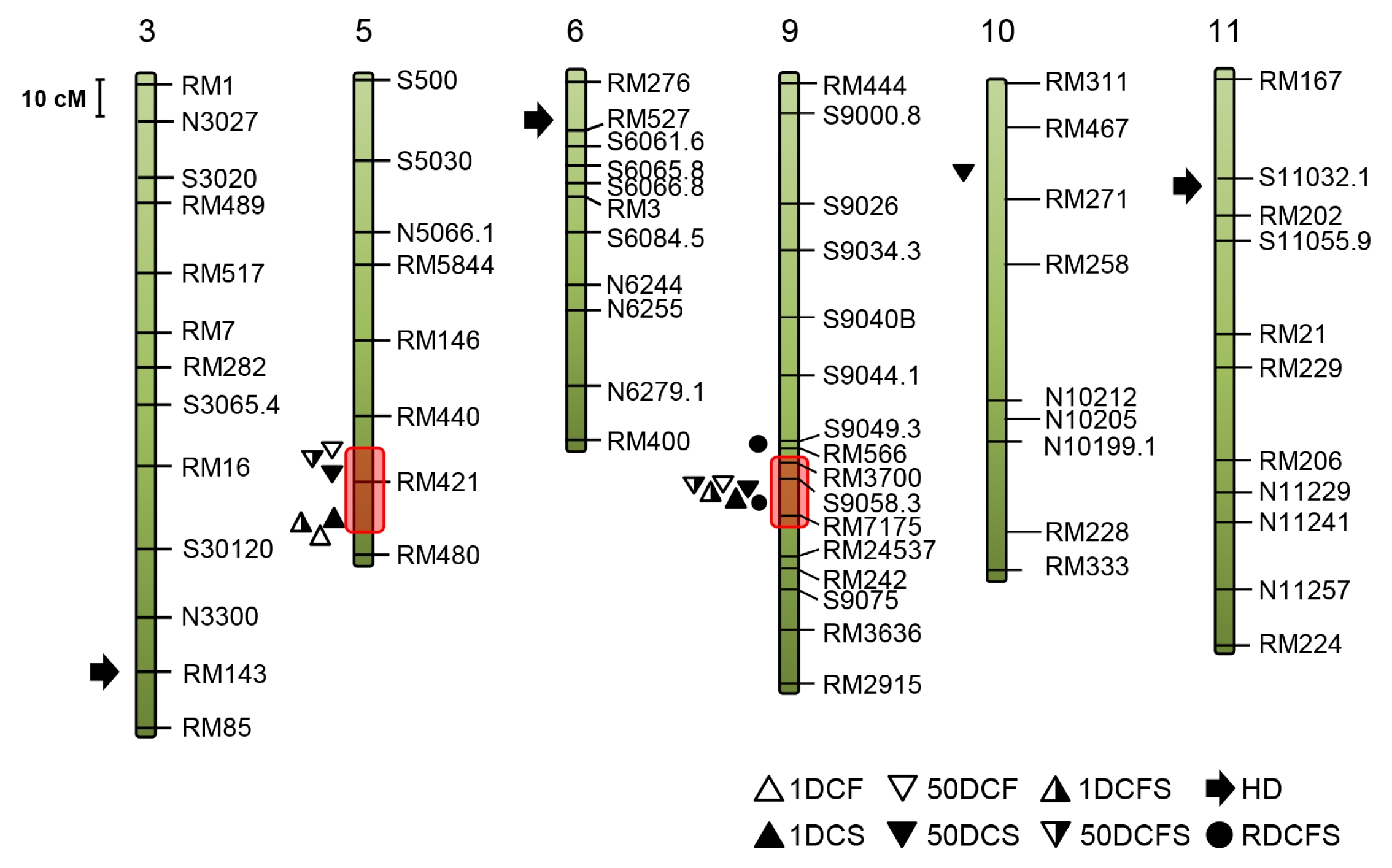
Fig. 3Development of heterogeneous inbred family-near isogenic lines (H-NILs) for 1Dcfs. The rows represent the genotypes of six F7 RILs that contain heterozygous genomic regions for the 1Dcfs9 QTL. Shaded regions indicate chromosomal segments that are homozygous SNU-SG1 (black), homozygous M23 (white), and heterozygous SNU-SG1/M23 (grey). 40 plants of F7:8 H-NILs derived from each F7 RILs were investigated for progeny tests (on the right). Maximum (Max) and minimum (Min) SPAD value for F7:8 H-NILs are shown with standard deviation (STV).
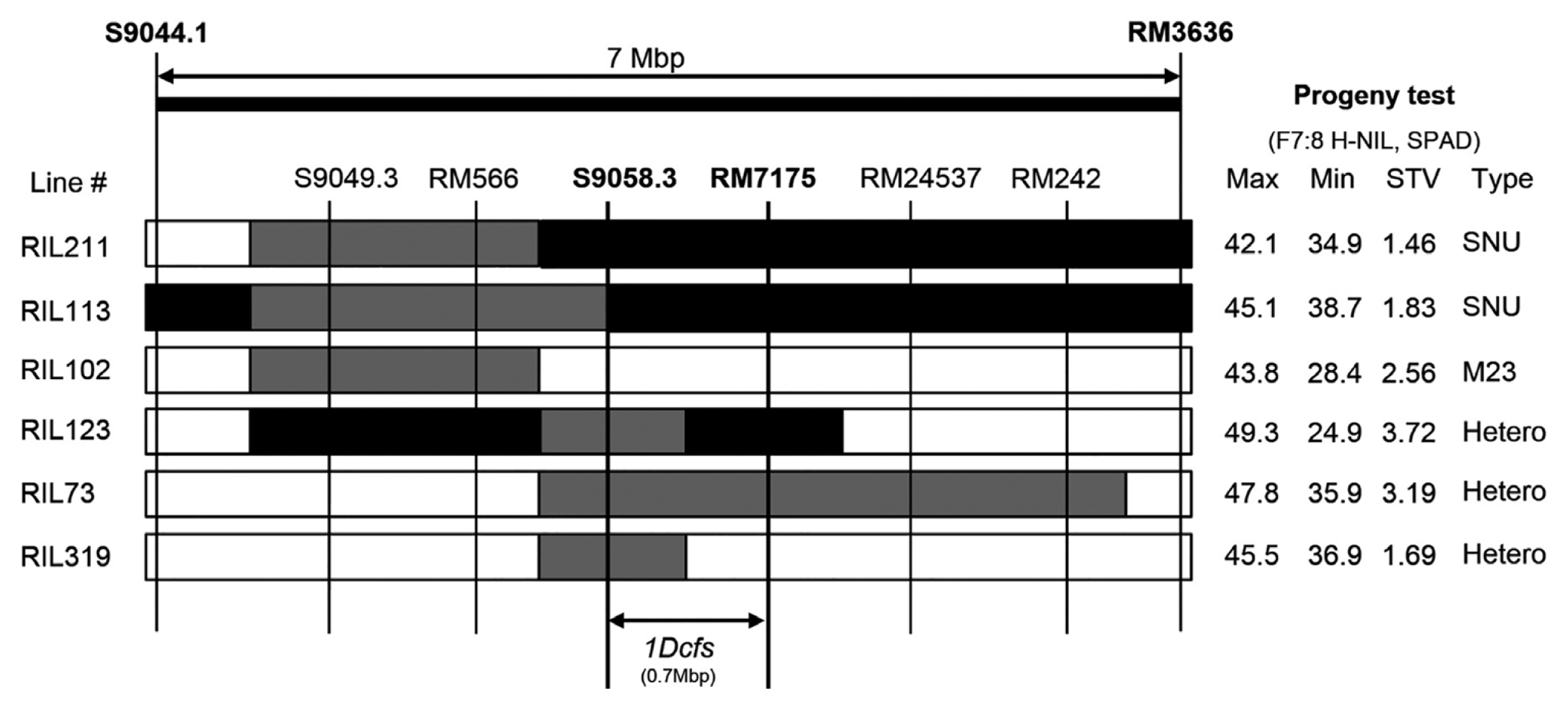
Fig. 4Genomic locations of two main-effect QTLs and their candidate genes. (A, B) Os06g16370 (A) and Os05g48270 (B) genes located at two main-effect QTLs, Hd6 and 1Dcfs5, respectively. (C, D) Gene structures and natural variations of Os06g16370 (C) and Os05g48270 (D). Sequence variations between SNU-SG1 and M23 are indicated. Black dots represent the absence of the corresponding bases. (E) Sequence alignments of the cytochrome b561 reductase domains of SNU-SG1, Nipponbare, Arabidopsis At3g61750, Sb09g027990, Horedeum vulgare and Zea mays ortholog. Black boxes indicate identical residues shared by at least three sequences; red box indicate complete conservation.
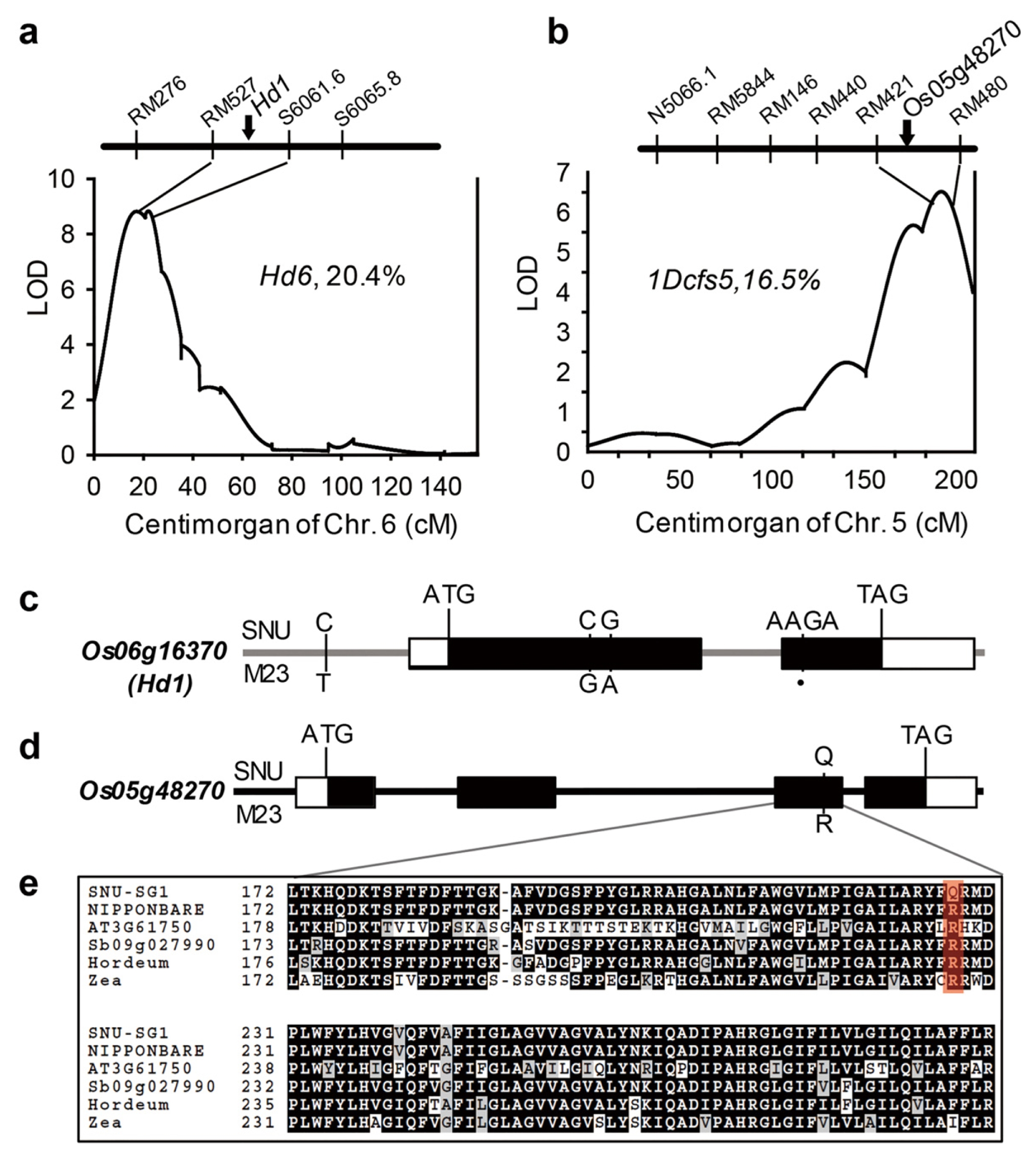
Table 1Descriptive statistics of the 13 traits for the parents and F7 RILs.
Table 1
|
Parents |
F7 RIL population |
|
|
|
Trait |
SNU-SG1 |
M23 |
Average |
Range |
SDz)
|
|
1DCF (SPAD) |
49.7 |
42.4 |
44.3 |
31.9–56.4 |
9.50 |
|
1DCS (SPAD) |
49.5 |
39.6 |
43.6 |
19.7–57.2 |
9.51 |
|
1DCFS (SPAD) |
49.6 |
41.1 |
44.0 |
26.3–55.8 |
9.37 |
|
50DCF (SPAD) |
34.6 |
13.4 |
26.2 |
1.8–54.9 |
12.9 |
|
50DCS (SPAD) |
35.3 |
12.9 |
23.4 |
1.9–55.7 |
12.1 |
|
50DCFS (SPAD) |
35.1 |
13.1 |
24.9 |
4–55.3 |
12.1 |
|
RDCFS (SPAD) |
0.71 |
0.32 |
0.56 |
0.10–0.94 |
0.23 |
|
Heading date (Days) |
105 |
113 |
105 |
88 – 134 |
9.13 |
Table 2Correlations of the eight traits analyzed in the F7 RIL population.
Table 2
|
1DCF |
1DCS |
1DCFS |
50DCF |
50DCS |
50DCFS |
RDCFS |
|
1DCS |
0.81**
|
|
|
|
|
|
|
|
1DCFS |
0.95**
|
0.96**
|
|
|
|
|
|
|
50DCF |
0.55**
|
0.50**
|
0.56**
|
|
|
|
|
|
50DCS |
0.56**
|
0.52**
|
0.57**
|
0.85**
|
|
|
|
|
50DCFS |
0.57**
|
0.54**
|
0.59**
|
0.97**
|
0.96**
|
|
|
|
RDCFS |
0.38**
|
0.35**
|
0.39**
|
0.95**
|
0.92**
|
0.97**
|
|
|
HD |
−0.29**
|
−0.21**
|
−0.26**
|
−043**
|
−0.44**
|
−0.44**
|
−0.42**
|
Table 3QTL identification for the 13 agronomic traits by composite interval mapping using 178 F7 RILs.
Table 3
|
QTL |
Chr.z)
|
Markery)
|
Ax)
|
LOD |
LOD peak (cM) |
R2 w) (%) |
95% CIv) (cM) |
Peru)
|
|
1 Degree of chlorophyll content of flag leaf
|
|
1Dcf5
|
5 |
RM421-RM480 |
−2.94 |
5.69 |
192.6 |
14.1 |
168.3–206.2 |
2.99 |
|
1Dcf9
|
9 |
RM3700-S9058.3 |
1.81 |
4,35 |
127.2 |
11.3 |
118.6–159.6 |
3.03 |
|
1 Degree of chlorophyll content of second leaf
|
|
1Dcs5
|
5 |
RM421-RM480 |
−3.07 |
6.11 |
190.8 |
15.2 |
164.3–203.8 |
2.89 |
|
1Dcs9
|
9 |
S9058.3-RM7175 |
2.07 |
4.51 |
136.6 |
11.7 |
115.8–153.7 |
2.92 |
|
1 Degree of chlorophyll content of flag and second leaf
|
|
1Dcfs5
|
5 |
RM421-RM480 |
−3.01 |
6.52 |
191.8 |
16.5 |
167.1–204.3 |
2.69 |
|
1Dcfs9
|
9 |
S9058.3-RM7175 |
2.01 |
4.88 |
137.5 |
12.0 |
121.8–155.0 |
2.83 |
|
50 Degree of chlorophyll content of flag leaf
|
|
50Dcf5
|
5 |
RM440-RM421 |
−5.20 |
3.78 |
175.5 |
9.2 |
157.2–202.0 |
2.74 |
|
50Dcf9
|
9 |
S9058.3-RM7175 |
4.44 |
3.97 |
137.3 |
10.7 |
108.4–164.1 |
2.76 |
|
50Dcf10
|
10 |
RM467-RM272 |
−4.36 |
3.43 |
19.3 |
9.8 |
1.7–36.2 |
2.87 |
|
50 Degree of chlorophyll content of second leaf
|
|
50Dcs5
|
5 |
RM421 |
−3.64 |
3.15 |
184.2 |
8.7 |
158.1–204.0 |
2.77 |
|
50Dcs9
|
9 |
S9058.3-RM7175 |
4.14 |
3.93 |
137.3 |
10.1 |
109.2–158.3 |
2.71 |
|
50 Degree of chlorophyll content of flag and second leaf
|
|
50Dcfs5
|
5 |
RM440-RM421 |
−4.45 |
3.71 |
177.3 |
9.5 |
158.6–202.0 |
2.76 |
|
50Dcfs9
|
9 |
S9058.3-RM7175 |
4.30 |
4.28 |
137.3 |
11.8 |
110.6–154.4 |
2.87 |
|
Heading date
|
|
Hd3
|
3 |
RM143 |
14.7 |
2.80 |
266.4 |
6.9 |
247.9–284.5 |
2.79 |
|
Hd6
|
6 |
RM527 |
6.49 |
8.83 |
17.4 |
20.4 |
10.8–25.9 |
2.76 |
|
Hd11
|
11 |
S11055.9-RM21 |
2.62 |
2.99 |
56.5 |
7.4 |
30.4–78.8 |
2.62 |
|
Relative degree of chlorophyll content of flag and second leaf
|
|
Rdsfs9-1
|
9 |
RM566 |
0.07 |
3.01 |
122.2 |
8.3 |
105.4–159.7 |
2.83 |
|
Rdcfs9-2
|
9 |
S9058.3-RM7175 |
0.07 |
3.17 |
137.4 |
8.8 |
105.4–158.7 |
2.83 |
Table 4Comparison of the QTLs identified in current and previous studies using the mapping populations derived from crosses of indica and japonica cultivars.
Table 4
|
QTL of this study |
QTL of previous studies |
|
|
|
QTL |
Physical Position(MB) |
QTL |
Marker region |
Physical position (Mb) |
PTz)
|
PSy)
|
Mapping parents japonica (J)/indica (I) |
Reference |
|
Hd3x)
|
30.0–33.1 |
Hd6
|
R3226 |
33.2 |
BC4F2
|
100 |
Nipponbare(J)/Kasalath(I) |
Yamamoto et al. (2000)
|
|
Hd6
|
9.8–11.1 |
Hd-1
|
R1679 |
8.9 |
F2
|
186 |
Nipponbare(J)/Kasalath(I) |
Yano et al. (1997)
|
|
|
qHd6-1
|
S2539-R2123 |
9.3–11.6 |
BIL |
182 |
Koshihikari(J)/Kasalath(I) |
Zhang et al. (2008)
|
|
Hd11
|
11.9–16.0 |
qHd-11
|
RM202-RM287 |
9.0–16.7 |
RIL |
184 |
Koshihikari(J)/Guichao2(I) |
Zhang et al. (2008)
|
|
1Dcfs5
|
21.0–24.0 |
dcfs5
|
RM440-RM430 |
21.0–22.5 |
RIL |
92 |
SNU-SG1(J)/M23(I) |
Yoo et al. (2007)
|
|
50Dcf10
|
15.0–18.0 |
rdgf10
|
RM269b-RM304 |
16.0–18.0 |
DHw)
|
190 |
Zhenshan97(I)/Wuyujing(J) |
Jiang et al. (2004)
|
|
RDCFS 9-1
|
15.4–15.9 |
Rdgf9
|
MRG2533-RM257 |
15.6–16.3 |
DHw)
|
190 |
Zhenshan97(I)/Wuyujing(J) |
Jiang et al. (2004)
|
|
|
qCCAJ-9
|
RM242-RM553 |
15.9–16.7 |
DHw)
|
135 |
IR36(I)/Nekken-2(J) |
Abdelkhalik et al. (2005)
|
|
|
dcfs9
|
RM566-S09040 |
14.7–15.8 |
RIL |
92 |
SNU-SG1(J)/M23(I) |
Yoo et al. (2007)
|
|
|
csfl9
|
RM434-RM242 |
15.5–18.8 |
RIL |
126 |
SNU-SG1(J)/Suwon490(I) |
Fu et al. (2011)
|
|
|
csfl9
|
RM434-RM242 |
15.5–18.8 |
RIL |
126 |
SNU-SG1(J)/Andabyeo(I) |
Fu et al. (2011)
|
Table 5List of 16 candidate genes for the 10 main-effect QTLs.
Table 5
|
QTL |
Gene ID |
Protein |
Description |
Type |
|
Hd6
|
Os06g16370
|
F.S.z)
|
HD1 |
My)
|
|
Hd6
|
Os06g17500 |
G133R |
expressed protein, Somatotropin_CS |
Sx)
|
|
Hd6
|
Os06g17510 |
I173V |
hypothetical protein |
S |
|
Hd6
|
Os06g17560 |
>6 A.A |
cadmium tolerance factor, putative |
S |
|
Hd6
|
Os06g17880 |
>6 A.A |
NBS-LRR disease resistance protein, putative |
S |
|
Hd6
|
Os06g17900 |
>6 A.A |
disease resistance protein RPM1, putative |
S |
|
Hd6
|
Os06g17910 |
>6 A.A |
NBS-LRR disease resistance protein |
S |
|
Hd6
|
Os06g17920 |
>6 A.A |
NBS-LRR disease resistance protein |
S |
|
Hd6
|
Os06g17930 |
>6 A.A |
NBS-LRR disease resistance protein |
S |
|
Os05g34380 |
A491V |
Obtusifoliol 14-alpha demethylase |
S |
|
1Dcf, 1Dcs, 1Dcfs, 50Dcf, 50Dcs, 50Dcfs
|
Os05g39540
|
Y231@ |
Zinc/iron permease family protein. |
S |
|
Os05g48270
|
R227Q |
DOMON related domain containing protein. |
S |
|
1Dcs, 1Dcf, 1Dcfs, 50Dcf, 50Dcs, 50Dcfs, Rdcfs
|
Os09g26530
|
L192S |
expressed protein / Ser/Cys_Pept_Trypsin-like |
S |
|
Os09g26554
|
C58F |
expressed protein / Ser/Cys_Pept_Trypsin-like |
S |
|
50Dcf10
|
Os10g28180 |
F.S. |
hAT dimerisation domain containing protein |
S |
|
Os10g31820 |
&823L |
fluG, putative, Similar to Nodulin 6l |
M |
Table 6List of seven strong candidate genes for the 10 main-effect QTLs.
Table 6
|
QTL |
Gene ID |
Protein |
Description |
Otholog |
|
Hd6
|
Os06g16370 |
F.S.z)
|
HD1 |
AT5G15840 |
|
1Dcf, 1Dcs, 1Dcfs, 50Dcf, 50Dcs, 50Dcfs
|
Os05g39540 |
Y231@ |
Zinc/iron permease family protein. |
AT1G05300 |
|
Os05g48270 |
R227Q |
DOMON related domain containing protein. |
Bradi2g17070 |
|
1Dcs, 1Dcf, 1Dcfs, 50Dcf,50Dcs, 50Dcfs, Rdcfs
|
Os09g26530 |
L192S |
expressed protein / Ser/Cys_Pept_Trypsin-like |
- |
|
Os09g26554 |
C58F |
expressed protein / Ser/Cys_Pept_Trypsin-like |
- |
|
50Dcf10
|
Os10g28180 |
F.S. |
hAT dimerisation domain containing protein |
- |
|
Os10g31820 |
&823L |
fluG, putative, Similar to Nodulin 6l |
AT3G53180 |
References
- Abdelkhalik AF, Shishido R, Nomura K, Ikehashi H. 2005. QTL-based analysis of leaf senescence in an indica/japonica hybrid in rice (Oryza Sativa L.). Theor Appl Genet. 110: 1226-1235.
- An H, Roussot C, Suárez-López P, Corbesier L, Vincent C, Piñeiro M, Coupland G. 2004. CONSTANS acts in the phloem to regulate a systemic signal that induces photoperiodic flowering of Arabidopsis. Develop. 131: 3615-3626.
- Doskočilová A, Plíhal O, Volc J, Chumová J, Kourová H, Halada P, Binarová P. 2011. A nodulin/glutamine synthetase-like fusion protein is implicated in the regulation of root morphogenesis and in signalling triggered by flagellin. Planta. 234: 459-476.
- D’Souza CA, Lee BN, Adams TH. 2001. Characterization of the role of the FluG protein in asexual development of Aspergillus nidulans. Genet. 158: 1027-1036.
- Dupuis J, Siegmund D. 1999. Statistical methods for mapping quantitative trait loci from a dense set of markers. Genet. 151: 373-386.
- Fu JD, Yan YF, Kim MY, Lee SH, Lee BW. 2011. Population-specific quantitative trait loci mapping for functional stay-green trait in rice (Oryza Sativa L.). Genome. 54: 235-243.
- Gentinetta E, Ceppl D, Lepori C, Perico G, Motto M, Salamini F. 1986. A major gene for delayed senescence in maize. Pattern of photosynthates accumulation and inheritance. Plant Breed. 97: 193-203.
- Han SH, Yoo SC, Lee BD, An G, Paek NC. 2015. Rice FLAVIN-BINDING, KELCH REPEAT, F-BOX 1 (OsFKF1) promotes flowering independent of photoperiod. Plant cell environ.
- Hayama R, Yokoi S, Tamaki S, Yano M, Shimamoto K. 2003. Adaptation of photoperiodic control pathways produces short-day flowering in rice. Nature. 422: 719-722.
- Jiang GH, He YQ, Xu CG, Li XH, Zhang Q. 2004. The genetic basis of stay-green in rice analyzed in a population of doubled haploid lines derived from an indica by japonica cross. Theor Appl Genet. 108: 688-698.
- Kojima S, Takahashi Y, Kobayashi Y, Monna L, Sasaki T, Araki T, Yano M. 2002. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 43: 1096-1105.
- Lander ES, Botstein D. 1989. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 121: 185-199.
- Lasat MM, Pence NS, Garvin DF, Ebbs SD, Kochian LV. 2000. Molecular physiology of zinc transport in the Zn hyperaccumulator Thlaspi caerulescens. J Exp Bot. 51: 71-79.
- Lee SH, Sakuraba Y, Lee T, Kim KW, An G, Lee HY, Paek NC. 2014. Mutation of Oryza sativa CORONATINE INSENSITIVE 1b (OsCOI1b) delays leaf senescence. J Integr Plant Biol.
- Lim JH, Yang HJ, Jung KH, Yoo SC, Paek NC. 2014. Quantitative trait locus mapping and candidate gene analysis for plant architecture traits using whole genome re-sequencing in rice. Mol Cells. 37: 149-160.
- Mae T. 1997. Physiological nitrogen efficiency in rice: nitrogen utilization, photosynthesis, and yield potential In Plant nutrition for sustainable food production and environment. Springer. Netherland: pp. 51-60.
- Neto EC, Keller MP, Broman AF, Attie AD, Jansen RC, Broman KW, Yandell BS. 2012. Quantile-based permutation thresholds for quantitative trait loci hotspots. Genetics. 191: 1355-1365.
- Park JH, Lee BW. 2003. Photosynthetic characteristics of rice cultivars with depending on leaf senescence during grain filling. Kor J Crop Sci.
- Park SY, Yu JW, Park JS, Li J, Yoo SC, Lee NY, Paek NC. 2007. The senescence-induced staygreen protein regulates chlorophyll degradation. Plant Cell. 19: 1649-1664.
- Pierce RO, Knowles PF, Phillips D. 1984. Inheritance of delayed leaf senescence in soybean. Crop Sci. 24: 515-517.
- Putterill J, Robson F, Lee K, Simon R, Coupland G. 1995. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell. 80: 847-857.
- International Rice Genome Sequencing Project.2005. The map-based sequence of the rice genome. 2005.
- Routaboul JM, Dubos C, Beck G, Marquis C, Bidzinski P, Loudet O, Lepiniec L. 2012. Metabolite profiling and quantitative genetics of natural variation for flavonoids in Arabidopsis. J Exp Bot. 63: 3749-3764.
- Schittenhelm S, Menge HU, Oldenburg E. 2004. Photosynthesis, carbohydrate metabolism, and yield of phyto-chrome-B-overexpressing potatoes under different light regimes. Crop Sci. 44: 131-143.
- Spano G, Di N, Perrotta C, Platani C, Ronga G, Lawlor DW, Shewry PR. 2003. Physiological characterization of ‘stay green’ mutants in durum wheat. J Exp Bot. 54: 1415-1420.
- Thomas H, Howarth CJ. 2000. Five ways to stay green. J Exp Bot. 5: 329-337.
- Tuinstra MR, Ejeta G, Goldsbrough PB. 1997. Heterogeneous inbred family (HIF) analysis: a method for developing near-isogenic lines that differ at quantitative trait loci. Theor Appl Genet. 95: 1005-1011.
- Walulu RS, Rosenow DT, Wester DB, Nguyen HT. 1994. Inheritance of the stay green trait in sorghum. Crop Sci. 3: 970-972.
- Weber M, Harada E, Vess C, Roepenack-Lahaye EV, Clemens S. 2004. Comparative microarray analysis of Arabidopsis thaliana and Arabidopsis halleri roots identifies nicotianamine synthase, a ZIP transporter and other genes as potential metal hyperaccumulation factors. Plant J. 37: 269-281.
- Yamamoto T, Lin H, Sasaki T, Yano M. 2000. Identification of heading date quantitative trait locus Hd6 and characterization of its epistatic interactions with Hd2 in rice using advanced backcross progeny. Genet. 154: 885-891.
- Yano M, Harushima Y, Nagamura Y, Kurata N, Minobe Y, Sasaki T. 1997. Identification of quantitative trait loci controlling heading date in rice using a high-density linkage map. Theor Appl Genet. 95: 1025-1032.
- Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Sasaki T. 2000. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell. 12: 2473-2483.
- Yoo S, Cho S, Zhang H, Paik H, Lee C, Li J, Paek NC. 2007. Quantitative trait loci associated with functional stay-green SNU-SG1 in rice. Mol Cells. 24: 83-94.
- Bing Y, Wei-Ya XUE, Li JLUO, Yong-Zhong XING. 2006. QTL analysis for flag leaf characteristics and their relationships with yield and yield traits in rice. Acta Genet Sinica. 33: 824-832.
- Zhang L, Li H, Li Z, Wang J. 2008. Interactions between markers can be caused by the dominance effect of quantitative trait loci. Genet. 180: 1177-1190.