Abstract
Plant breeding programs are often used to improve varieties through creating diverse agronomic traits. During a breeding program, a lot of genetic diversities are created in the genome after different generations through homologous recombination. Genome sequencing technology has revolutionized the discovery of genes and molecular markers associated with diverse agronomic traits in crop improvement programs. Genomic research is now in the peak of success, thus creating new opportunities for crop improvement modern sequencing technology is now capable of sequencing thousands to millions of bases per run. Modern sequencing technologies enable the sequencing of different cultivars with small to complex genomes at a reasonable time and cost. These massive data can be used to identify important agronomic traits of crops such as fruit color, size, ripening, flowering time adaptation, grain yield, and quality maintenance. In addition, they can be used to develop crop varieties. This mini-review is focused on the role of genome sequencing in genomic research and plant breeding for crop improvements.
-
Key words: Plant breeding, Genetic diversity, Homologous recombination, DNA sequencing, Molecular markers, Agronomic traits
INTRODUCTION
Plant breeding is a natural way to create genetic variation among individuals of species in order to obtain desired characteristics. It is important to ensure that the world food demand is met by developing new varieties with improved crop qualities, including high yield, tolerance to various environmental stresses (drought, cold, salinity, flood, etc.), and resistance to various insects, fungi, bacteria, and viruses. Breeding programs can develop crop varieties with these qualities. One major technique in plant breeding is by crossing between two closely related individuals to produce new crop varieties or lines with desirable properties. A successful breeding program largely depends on homologous recombination to exchange genetic information between chromosomes during meiosis, thus creating genetic diversity and hybrid plants with desired characteristics. These genomic changes in individuals could be associated with phenotype changes, including different colors of plants or fruits, size, grain yields, tolerance to various stresses, male sterility in plants, and different disease resistance. Phenotypic variations through domestication from one region to other regions are also associated with different agricultural species. Analysis of DNA sequence variations in candidate genes based on morphological phenotypes is a good way to identify causal genes for these traits. Many studies have reported genome-wide evaluations for different breeding varieties or lines of plants with strong genetic diversity across different chromosomal regions in association with different phenotypic changes (
Lam et al. 2010;
Riedelsheimer et al. 2012;
Causse et al. 2013;
Chen et al. 2014;
Xu et al. 2014).
Modern sequencing technology is a great tool for identifying the genetic diversity in hybrid plants, especially for discovering genes and developing molecular markers associated with diverse agronomic traits of crops. Recent advances in genome sequencing technology have made it possible to sequence thousands to millions of bases per run within a short time at a low cost so that millions of molecular markers can be developed for different crop species. In addition, they can be used to identify agronomically important genes. Complete reference genome sequences are now available for several agronomic important crop species, making it relatively easy to rapidly identify candidate genes or detect genetic variation during breeding events (
Goff et al. 2002;
Yu et al. 2002;
Velasco et al. 2007;
Velasco et al. 2010;
Potato Genome Sequencing Consortium et al. 2011;
Mayer et al. 2012;
Tomato Genome Consortium 2012;
Hirakawa et al. 2014). Discovery of millions of novel molecular markers for important crop plants has revolutionized plant genomic research. These markers can be regularly used in plant breeding programs for quick development of crops, analysis of genetic variations, and identification of different varieties or cultivars or genes with important agronomic traits.
MODERN SEQUENCING TECHNOLOGY
The sequencing technology was invented at the early 1970s. The first complete genome of Bacteriophage
phi X174 (genome size: 5,386 bases) was sequenced in 1977 through Sanger method (
Wu and Kaiser 1968;
Gilbert and Maxam 1973;
Sanger and Coulson 1975;
Sanger et al. 1977a,
1977b). Sanger method is a sequencing-by-synthesis method. It depends on a combination of deoxy- and dideoxy-labeled chain terminator nucleotides (
Sanger et al. 1977a). Since then, the sequencing technology has developed day by day for sequencing whole or partial genomes. Current Sanger method can read about 6 Mb per day with read length of approximately 800 nucleotides. However, the cost is very high (around 500$ per Mb). Sanger-based method is only applicable for small size genomes or samples. Over the last decade, new sequencing technologies have become available (
Margulies et al. 2005;
Shendure et al. 2005;
Bentley et al. 2008;
Harris et al. 2008;
Korlach et al. 2008;
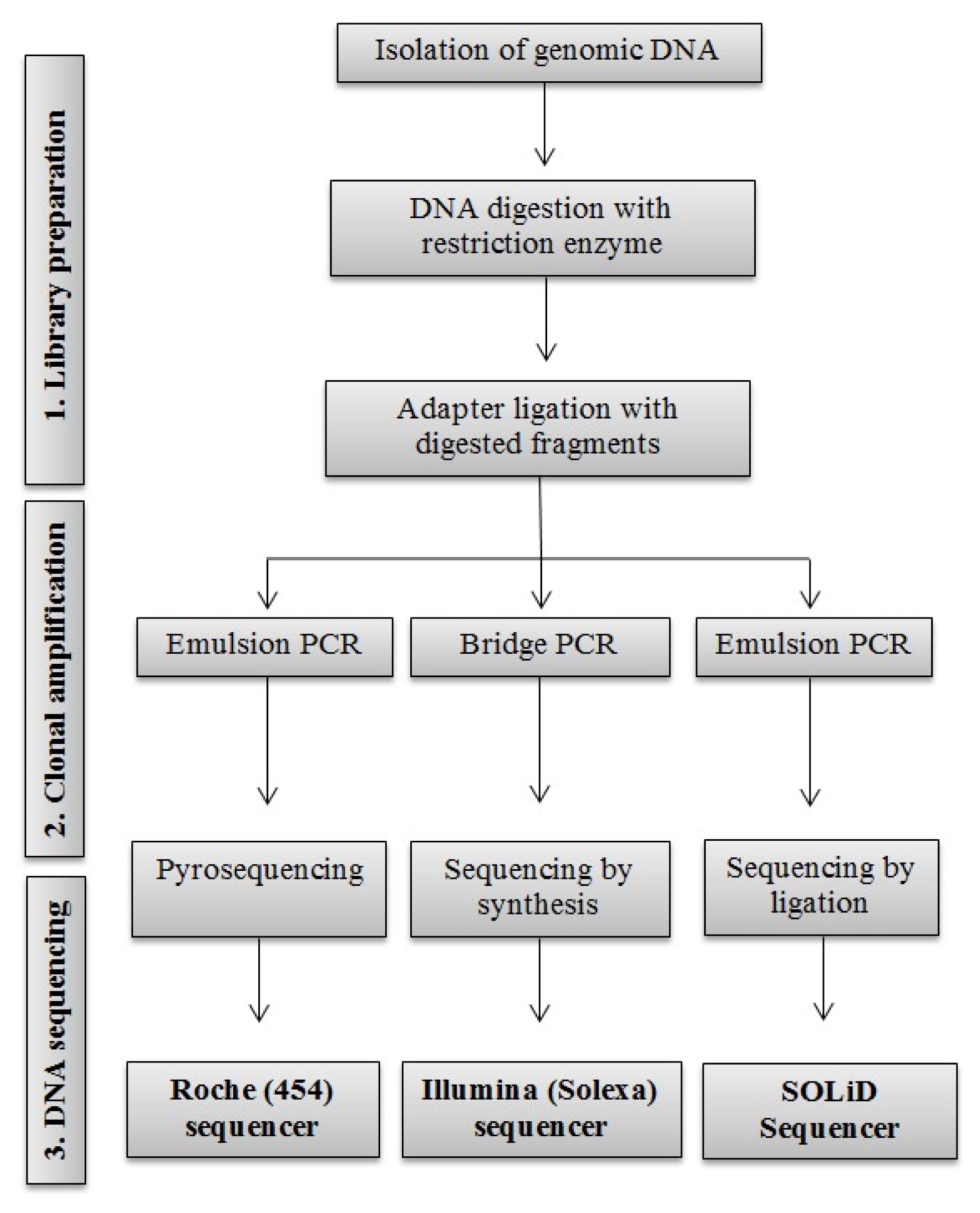
Clarke et al. 2009). They have outperformed Sanger-based sequencing in throughput and the overall cost, with approximately 100 to 1,000 times faster in daily throughput at price 25 to 1,000 times cheaper than Sanger-based method. In addition, these methods are applicable for any type of genomes, including complex genomes and many samples. Because of these positive changes in sequencing techniques, different companies and researchers have use the term of ‘next-generation sequencing’ for these newly developed high-throughput sequencing technologies in the last decade. Three main next-generation sequencing techniques with their throughput and relative cost will be focused in this review. A schematic representation of the three DNA sequencing methods is shown in
Fig. 1.
ROCHE (454) SEQUENCING
Roche (454) sequencing was launched to the market in 2005 as the first high-throughput next-generation DNA sequencing technology. It is based on pyrosequencing also known as sequencing by-synthesis. In this method, single fragments of DNA are hybridized to an array of capture bead that contains all necessary reagents for polymerase chain reaction (PCR) amplification of individually bound template. The bead also contains enzymes so that fluorescence can be generated through consumption of inorganic phosphate. During sequencing, one pyro-phosphate is released from each nucleotide. The released pyrophosphate is detected by an enzymatic luminometric inorganic pyrophosphate detection assay through creating a light signal during the transformation of PPi into adenosine triphosphate (ATP) by ATP sulfurylase (
Ronaghi et al. 1996). The current Roche (454) pyrosequencing method can generate up to 700 Mb in a 23-hour run with an average read length of around 750 nucleotides. The cost is approximately 9$ per Mb. The overall error rate is approximately 1% (
Glenn 2011).
ILLUMINA SEQUENCING
After the launch of Roche (454) sequencing, another high-throughput reversible terminator sequencing technology that was released to the market was Illumina (Solexa) sequencing using the sequencing-by-synthesis concept (
Bentley et al. 2008;
Turcatti et al. 2008). Like Roche (454), Illumina sequencing technology also needs to prepare DNA library through genomic DNA digestion and adapters ligation so that DNAs can be amplified and immobilized for sequencing (
Fedurco et al. 2006;
Bentley et al. 2008). Two different adapters are added to the 5′ and 3′ ends of all digested DNA fragments. After adapter ligation, primers are used to amplify the sequences through bridge amplification system. DNA molecules are then sequenced by flooding the flow cell with a new type of cleavable fluorescent nucleotides that can emit light with reagents necessary for DNA polymerization (
Turcatti et al. 2008). During synthesis of the complementary strand, the fluorescent molecule is excited by a laser to emit light that is different in color for each of the four bases. Illumina sequencing is a paired-end sequencing system that can generate sequence reads from both end of DNA fragments. Illumina sequencing is more advanced than Roche (454) pyrosequencing with high-throughput and less cost. HiSeq2500 sequencing instrument from Illumina can read around 600 Gb per run with an error rate of 0.1% and average read length of about 100 nucleotides at cost of approximately 23,500$ per run (
Glenn 2011).
APPLIED BIOSYSTEMS (SOLiD) SEQUENCING
SOLiD sequencing platform was invented as the third high-throughput next generation sequencing system. It was jointly developed by Harvard Medical School and Howard Hughes Medical Institute (
Shendure et al. 2005). It was commercially released to the market in 2007. SOLiD system is very similar to Roche (454) in library preparation where clonal bead populations are made in micro-reactors that contain beads, DNA template, primers, and PCR reagents. For amplification, they also use the same emulsion PCR as in Roche (454). On beads, DNA templates are modified by adapter binding. A primer is annealed to an adapter of the DNA template. Amplification is achieved by pumping a mixture of fluorescently tagged oligonucleotides into flow cell. When oligonucleotide matches the template sequence and binds with the primer, a color will be shown and a camera will capture different colors (four colors for four bases). After collecting images, the fluorescent tag is removed and a new set of oligonucleotides are injected into the flow cell to start the next round of DNA ligation (
Shendure et al. 2008). This method is called sequencing-by-ligation method. The SOLiD platform can yield up to 1,410 million paired-end reads per run with 0.01% error rate. The average read length is 75+35 nucleotides with an approximate cost of 10,500$ per run (
Glenn 2011).
GENOME SEQUENCING PROJECTS FOR CROP PLANTS
The first complete plant genome (the model plant
Arabidopsis thaliana) was successfully sequenced in 2000 using bacterial artificial chromosome (BAC)-based approach. Its genome size was approximately 125 Mb (
The Arabidopsis Genome Initiative 2000). It was a great achievement in plant genomic research. Sequencing of the rice genome using the same technique was another milestone for crop research because it was the first sequenced crop genome with a size of 389 Mb, which was about three times larger than model plant
Arabidopsis (
Goff et al. 2002;
Yu et al. 2002;
International Rice Genome Sequencing Project 2005). Recently, crop genome sequencing projects are rapidly increasing with the development of new high-throughput sequencing technology. In 2007, the grape genome of 504 Mbp was sequenced using both Sanger and Roche (454) methods (
Velasco et al. 2007). The complete genome of
Theobroma cacao was sequenced in 2011 using Roche (454), Illumina and Sanger sequencing technologies (
Argout et al. 2011). To obtain the genome of apple, a combination of Sanger and Roche (454) sequencing was used (
Velasco et al. 2010). In 2012, a combination of three technologies (Sanger, Roche, and Illumina) was used to survey the genome of banana (
D’Hont et al. 2012). In 2013, the genome of oil palm at 1.8 Gb was sequenced using a combination of Roche (454) and Sanger BAC-end sequencing techniques (
Singh et al. 2013). The genome of
Brassica rapa was sequenced and analyzed in 2011 using Illumina GAII sequencer (
Brassica rapa Genome Sequencing Project;
Wang et al. 2011). The genome of cucumber was assembled through a hybrid strategy consisting of Sanger, Illumina (Solexa), end sequenced fosmid, and BAC libraries (
Huang et al. 2009a). The whole genome of maize at 2.3 Gb was first sequenced in 2009 using a minimum tiling path of BACs and fosmid clones (
Schnable et al. 2009). The genome of soybean at 1.1 Gb was first sequenced in 2010 using whole-genome shotgun approach with Sanger sequencing protocols (
Schmutz et al. 2010). The genome of potato at 850 Mb was revealed by the Potato Genome Sequencing Consortium
et al. in 2011 using a combined approach of two next-generation sequencing (NGS) platforms of Roche pyrosequencing and Illumina genome analyzer as well as the conventional Sanger sequencing technologies. Tomato Genome Consortium has sequenced and assembled the complete genome of tomato in 2012 using Roche (454) and Sanger paired-end reads (
Tomato Genome Consortium 2012). The genome of barley at around 5.1 Gb was first sequenced in 2012 using Sanger sequencing, Roche (454), and Illumina HiSeq 2000 (
Mayer et al. 2012). More than 50 plant genomes have been published in 2013, most of them are crop plants (
Michael and Jackson 2013). Since then, the number of plant genomes being sequenced is increasing day by day due to technological development. In 2014, the whole genome of eggplant was sequenced and assembled using Illumina HiSeq 2000 and Roche (454) GS FLX (
Hirakawa et al. 2014). The genome of cultivated cotton
Gossypium arboreum was sequenced in 2014 using Illumina sequencing platform (
Li et al. 2014). The size and hexaploid nature of bread wheat genome generated some problems in obtaining its genome sequences. However, The International Wheat Genome Sequencing Consortium revealed a chromosome-based draft genome sequence of hexaploid bread wheat in July 2014 (
International Wheat Genome Sequencing Consortium [IWGSC] 2014). Their vision is to generate high quality annotated and physical map-based integrated genome sequence of wheat within a short time. Some important crop plants with their genome sequencing databases are listed in
Table 1.
GENOME SEQUENCE INFORMATION DIRECTLY AFFECTS CROP RESEARCH AND IMPROVEMENTS
Sequenced genome of crops can be used to improve crop varieties in different ways, including the discovery of molecular markers, identification of agronomical important traits, and the transfer of these traits into elite varieties. Modern sequencing technology and sequenced genomes provide great opportunities to plant breeders for efficient marker assisted breeding, selection of quantitative trait locus (QTL), and management of genomic resources within a short time frame.
MOLECULAR MARKER DISCOVERY
The main advantage of genome sequence is the development of molecular markers that can be used to distinguish different species or varieties or cultivars to find possible elite variety. Molecular markers are developed based on genomic variations between individuals. The discovery of millions of novel genetic markers through genome sequencing is revolutionizing plant genomic research and crop improvements. Different types of molecular markers are now being widely used for analyzing genomic diversity in plants and for their development (
Henry 2001;
Phillips and Vasil 2001). Among these markers, single nucleotide polymorphisms (SNPs) are the most abundant ones due to their availability in the genomes of different populations. SNP markers are now widely used for the analysis of population structure, association mapping, selection of QTL, and/or evolutionary studies. Due to technological advances, thousands to millions of SNP markers are now available for each plant species. Whole genome sequencing is the best way to find genetic diversity and molecular markers in a population and gain a better understanding about the relationship between genotypic and phenotypic changes.
Chen et al. (2014) have identified more than 10,000,000 SNP loci from 801 re-sequenced rice varieties.
Xu et al. (2014) have found a total of 6,385,011 SNPs through re-sequencing 15 maize inbreds and compared them to maize B73 reference genome. Another study yielded a total of 4,290,679 unique SNPs from eight tomato lines when each genome was separately compared to the reference genome (
Causse et al. 2013).
QTL MAPPING
A QTL is a region of DNA or locus associated with a particular phenotype. QTLs typically contain genes that control the phenotype. Generally, a single phenotypic trait is determined by many genes. Therefore, many QTLs are associated with a single trait. By using complete genome sequencing data, it is possible to make QTL map so that it is easy to identify reasons behind phenotypic variations among individuals. High density molecular markers developed from sequenced genome can be used to rapidly map agronomical important traits and identify candidate genes within a region of interest.
Lam et al. (2010) have discovered a set of 205,614 tag SNPs by re-sequencing 31 wild and cultivated soybean genomes. They might be useful for QTL mapping and associated studies. The availability of rice genome sequence has made it easy to find different QTLs such as rice grain production containing cytokinin oxidase (
Ashikari et al. 2005). Likewise, it is possible to identify maize QTLs for biomass and bioenergy because of the availability of maize genome sequence (
Riedelsheimer et al. 2012).
ABIOTIC AND BIOTIC STRESS TOLERANCE VARIETY DEVELOPMENT
Abiotic stress is a stress to plants caused by non-living factors such as environmental and non-biological factors, including drought, flood, temperature, salt, radiation, and chemicals. On the other hand, biotic stress is caused by living organisms such as fungi, bacteria, viruses, insects, parasites, and weeds. Due to technological advances, researchers can now focus more on the development of crop varieties tolerant to different stresses. It is relatively easier to develop different stress tolerance varieties if the reference genome of important crop is available. Once the rice genome becomes available, researchers have identified a gene called
DST that is responsible for drought and salt tolerance in rice. Loss of this gene (
DST) function will enhance stomatal closure and decrease stomatal density, resulting in improved drought and salt tolerance in rice (
Huang et al. 2009b). Similarly, after the genome sequence of maize became available, candidate genes were identified for drought tolerance so that they could be used for the development of drought tolerance varieties (
Xu et al. 2014). In one study, more than 6,000 SNPs were found in drought stress related genes from two contrasting cultivars of Soybean: sensitive (BR 16) and tolerant (Embrapa 48). Among these SNPs, 165 were related to tolerance to abiotic stress (
Vidal et al. 2012).
Several genes involved in stress responses related to drought, heat, flooding, salinity, and pathogen defense were identified by transcriptome analysis for two switchgrass ecotypes: lowland variety AP13 and upland variety VS16 (
Fiedler et al. 2016). RNA binding proteins, Mei2-like, pumilio, and RRM domain containing proteins implicated in posttranscriptional gene regulation were also found (
Fiedler et al. 2016). Drought and heat are major concerns for wheat production as they can dramatically reduce grain yield. A QTL
qDHY.3BL was detected on chromosome 3BL of a double-haploid population after a cross between two wheat cultivars (RAC875 and Kukri), showing valuable source of abiotic stress tolerance (
Thomelin et al. 2016).
Sukumaran et al. (2016) developed and tested several physiological traits related to heat and drought stress in wheat. Genome-wide association mapping was done on a wheat association mapping initiative (WAMI) panel for these traits. Some QTLs associated with drought and heat tolerance in spring wheat were detected.
Li et al. (2016) performed genome-wide association studies (GWAS) of disease and drought-relative traits using whole genome re-sequencing data of Chickpea and found more than 800,000 SNPs from 64 Australian chickpea varieties, 4 Indian landraces, and 1 wild chickpea species. GWAS identified a 100 kb region on the chromosome 4 of chickpea that was significantly associated with ascochyta blight resistance, a fungus disease that could severely impact chickpea production in Australia and other regions of the world.
The wild grass species
Thinopyrum elongatum has been identified as a source of strong fusarium head blight (FHB) resistance. After being inserted into wheat by recombination FHB resistance wheat was developed (
Ouellet et al. 2016).
Allen et al. (2016) developed an enhanced fusarium crown rot resistance wheat cultivar by stimulating the ethylene-signaling pathway with ethylene biosynthetic precursor 1-aminocyclopropene-1carboxylic acid (ACC) and found associated genetic changes by RNA-Seq data analysis.
Mukhtar et al. (2009) found 230 SNPs in
Allene Oxide Synthase 2 gene of 184 tetraploid potato individuals associated with field resistance to late blight in populations of tetraploid potato cultivars. In another study,
Shi et al. (2015) identified three functional SNP in soybeans (two for Rhg1 locus and one for Rhg4 locus) responsible for soybean cyst nematode resistance.
FLOWERING TIME AND MATURATION TIME
Flowering time and maturation time are important agronomical traits involved in the adaptation to various geographical locations and crop yields. In a positive way, delayed flowering time prolongs the period of vegetative growth, thus increasing yields. But in a negative way, delayed flowering time might increase the susceptibility of plants to winter mortality, thus decreasing production. Therefore, controlling flowering time and maturation time of crops can help improve their production. Recent advances in genomic research have provided great opportunities to understand and regulate the flowering time of crops. Soybean genome was used to discover a maturity locus
E1 that could regulate the flowering time and maturity of soybean (
Xia et al. 2012). Likewise, potato genome sequence was used to identify a transcription factor that could regulate tuberization and plant life cycle length (
Kloosterman et al. 2013).
Grabowski et al. (2016) identified genic regions in the switchgrass genome that might play a role in controlling flowering time through GWAS. In another study, a pseudo-F2 population was generated by crossing two F1 siblings to genetically map flowering time genes in switchgrass (
Tornqvist et al. 2016).
Taylor et al. (2016) also discovered the genetic variation of flowering time and biomass in switchgrass mapping population and identified quantitative trait loci that could control the flowering time and biomass. Through GWAS in rice, a total of 32 new loci associated with the control of flowering time were isolated (Huang
et al. 2012).
Shaw et al. (2016) characterized the flowering locus
T2 (
FT2) in wheat and
Brachypodium. Two major external stimuli (temperature and light) were used by temperate cereals to coordinate flowering time with seasonal changes through vernalization and photoperiod pathways. These pathways converge on flowering locus
T1 (
FT1), a key gene in the induction of flowering whose deletion will delay flowering for less than one month. In that study,
FT2 homologs in temperate grasses
Brachypodium distachyon, wheat, and barley were characterized using transgenic and mutant approaches. It was found that
FT2 gene expression profiles were different from
FT1, suggesting that these two genes may have independent roles in the control of flowering in temperate cereals (
Shaw et al. 2016).
GRAIN QUALITY AND YIELDS
High yields and improved grain quality are the most desired traits in crop breeding programs. Once the rice genome sequence became available, it was immediately used to find major QTLs for rice grain production.
Ashikari et al. (2005) identified a QTL that could increase grain productivity of rice. Gene
Gn1a encodes
cytokinin oxidase/dehydrogenase (
OsCKX2), an enzyme that can degrade phytohormone cytokinin. Reduced expression of
OsCKX2 can cause cytokinin accumulation in inflorescence meristems and enhance the number of reproductive organs, resulting in increased grain yield. Huang
et al. (2012) recognized 10 grain-related loci through GWAS in rice. In one study, QTL for grain yield was analyzed in 243 F2 rice plants derived from a cross between IL28 and Ilpumbyeo. It was found that
qSPP6 and
qTGW6 alleles were involved in improving rice yields (
Ahn et al. 2016). Through GWAS in bread wheat (
Triticum aestivum), several putative loci on chromosomes 2A, 4A, and 2D were found to be associated with grain protein concentration (
Dao et al. 2016). In addition, several putative loci on chromosomes 1B, 2B, 3B, and 4B were found to be associated with for test weight (
Dao et al. 2016).
Guo et al. (2016) analyzed 54 yield-related traits in 200 wheat genotypes to genetically dissect assimilate partitioning between competing plant parts of wheat by GWAS and suggested that
VRN,
Ppd,
FT,
Rht, and
Q genes might be involved in the regulation of assimilate distribution between competing plant parts, an important strategy to improve wheat grain yield.
CONCLUSION
Recent advances in genome sequencing technology are radically changing genomic researches for plants, thus having a major impact on crop improvement. The success stories indicate that it is not so far to sequence complex and large crop genome and identify all genes including QTLs within a short time. New valuable whole-genome information will aid rapid genome wide association studies to make a link between genetic variation and agronomic traits for all crop species. The identification of QTL and molecular markers underlying these agronomic traits will speed up the breeding process and lead to improved varieties, including enhanced yield and quality, increased tolerance to adverse environmental conditions (such as severe drought, flood, cold, and extreme temperatures), and higher resistance to pathogens (such as bacteria, fungi, viruses, and insects).
Fig. 1
Overview of three high-throughput next generation sequencing systems.
PCR: polymerase chain reaction.
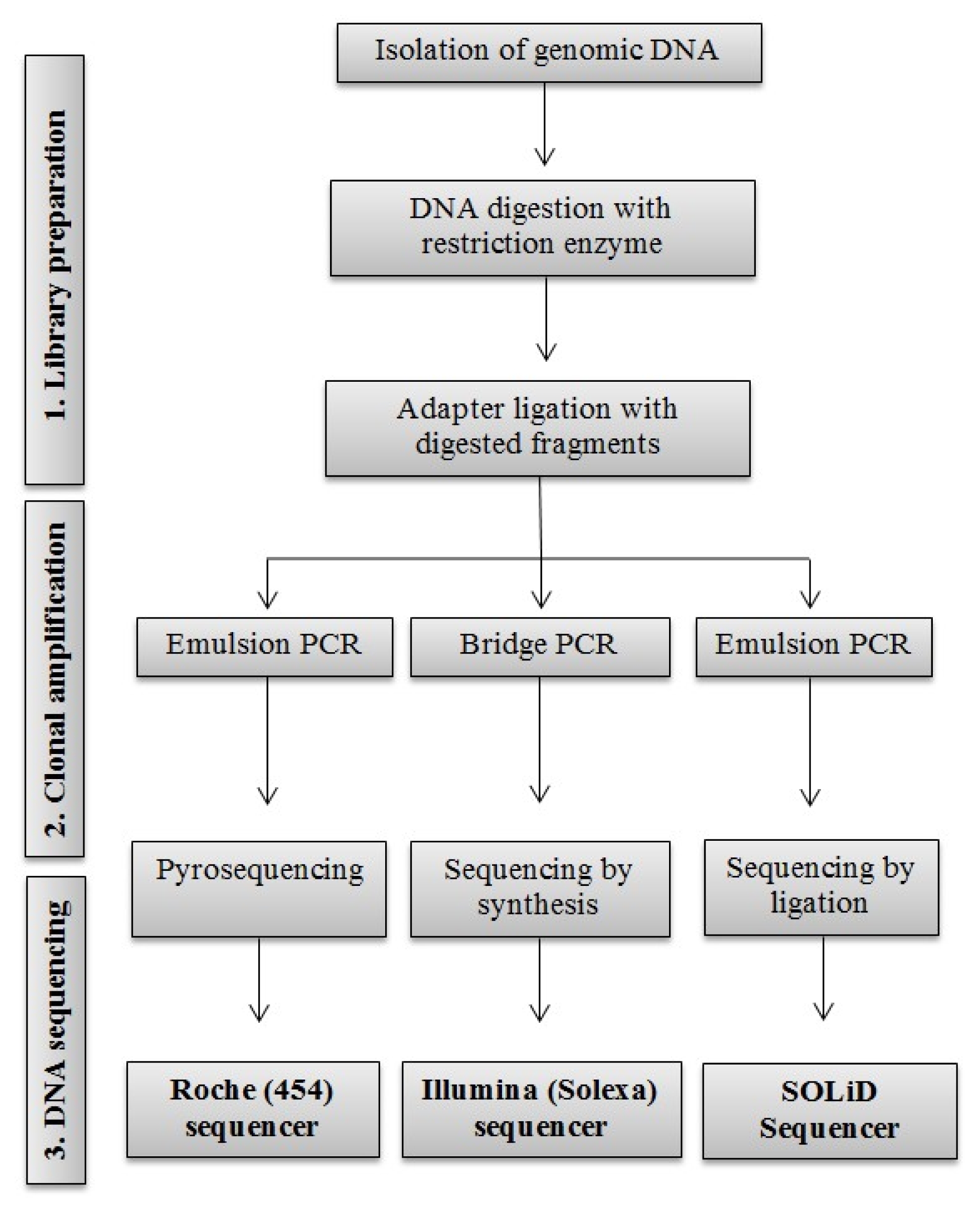
Table 1Some important crop plants with their genome sequencing databases.
Table 1
|
Crop |
Genome size |
Year of completion |
Technology used |
Reference |
|
Rice |
389 Mbz)
|
2002 |
BAC end sequencing |
, Yu et al. 2002
Goff et al. 2002
|
|
Grape |
504 Mb |
2007 |
Sanger and Roche (454) |
, Velasco et al. 2007
|
|
Maize |
2.3 Gb |
2009 |
Sanger |
, Schnable et al. 2009
|
|
Cucumber |
350 Mb |
2009 |
Sanger and Illumina |
, Huang et al. 2009a
|
|
Apple |
742 Mb |
2010 |
Sanger and Roche (454) |
, Velasco et al. 2010
|
|
Soybean |
1.1 Gb |
2010 |
Sanger |
, Schmutz et al. 2010
|
|
Potato |
850 Mb |
2011 |
Roche (454), Illumina and Sanger |
, Potato Genome Sequencing Consortium et al. 2011
|
|
Banana |
523 Mb |
2012 |
Sanger, Roche (454) and Illumina |
, D’Hont et al. 2012
|
|
Tomato |
950 Mb |
2012 |
Roche (454) and Sanger |
, Tomato Genome Consortium 2012
|
|
Barley |
5.1 Gb |
2012 |
Sanger, Roche (454) and Illumina |
, Mayer et al. 2012
|
|
Oil palm |
1.8 Gb |
2013 |
Roche (454) and Sanger |
, Singh et al. 2013
|
|
Eggplant |
1.1 Gb |
2014 |
Illumina and Roche (454) |
, Hirakawa et al. 2014
|
References
- Ahn SN, Kim DM, Lee HS, Kang JW, Yu YT. 2016. High-density mapping reveals a linkage of quantitative trait loci for grain-weight and spikelet number in rice. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0728.
- Allen S, Bhide K, Scofield S. 2016. The role of ethylene in PAMP-triggered immunity in Fusarium crown rot disease resistance in wheat. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0861.
- Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, et al. 2005. Cytokinin oxidase regulates rice grain production. Science. 309: 741-745.
- Argout X, Salse J, Aury JM, Guiltinan MJ, Droc G, Gouzy J, et al. 2011. The genome of Theobroma cacao. Nat Genet. 43: 101-108.
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 456: 53-59.
- Causse M, Desplat N, Pascual L, Le Paslier MC, Sauvage C, Bauchet G, et al. 2013. Whole genome resequencing in tomato reveals variation associated with introgression and breeding events. BMC Genomics. 14: 791
- Chen H, Xie W, He H, Yu H, Chen W, Li J, et al. 2014. A high-density SNP genotyping array for rice biology and molecular breeding. Mol Plant. 7: 541-553.
- Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. 2009. Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol. 4: 265-270.
- Dao HQ, Reid S, Harrington J, Byrne P. 2016. Genome-wide association study in bread wheat identifies loci for grain protein concentration and test weight. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0834.
- D’Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, Garsmeur O, et al. 2012. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 488: 213-217.
- Fedurco M, Romieu A, Williams S, Lawrence I, Turcatti G. 2006. BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies. Nucleic Acids Res. 34: e22
- Fiedler E, Ayyappan V, Sripathi VR, Saha MC, Thimmapuram J, Bhide K, et al. 2016. Transcriptome analysis of two switchgrass ecotypes: AP13 (lowland) and VS16 (upland). In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0017.
- Gilbert W, Maxam A. 1973. The nucleotide sequence of the lac operator. Proc Natl Acad Sci USA. 70: 3581-3584.
- Glenn TC. 2011. Field guide to next-generation DNA sequencers. Mol Ecol Res. 11: 759-769.
- Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science. 296: 92-100.
- Grabowski P, Evans J, Ramstein GP, Crisovan E, Vaillancourt B, Costich D, et al. 2016. Genome-wide associations with flowering time in switchgrass. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0001.
- Guo Z, Chen D, Ganal MW, Röder MS, Schnurbusch T. 2016. Genetic determinants of grain yield in wheat revealed by assimilate partitioning. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0832.
- Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, Braslavsky I, et al. 2008. Single-molecule DNA sequencing of a viral genome. Science. 320: 106-109.
- Henry RJ. 2001. Plant genotyping: the DNA fingerprinting of plants. CABI Publishing. Wallington, Oxon, New York.
- Hirakawa H, Shirasawa K, Miyatake K, Nunome T, Negoro S, Ohyama A, et al. 2014. Draft genome sequence of eggplant (Solanum melongena L.): the representative Solanum species indigenous to the old world. DNA Res. 21: 649-660.
- Huang X, Zhao Y, Wei X, Li C, Wang A, Zhao Q, et al. 2011. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet. 44: 32-39.
- Huang XY, Chao DY, Gao JP, Zhu MZ, Shi M, Lin HX. 2009b. A previously unknown zinc finger protein, DST, regulates drought and salt tolerance in rice via stomatal aperture control. Genes Dev. 23: 1805-1817.
- Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, et al. 2009a. The genome of the cucumber, Cucumis sativus L. Nat Genet. 41: 1275-1281.
- International Barley Genome Sequencing ConsortiumMayer KF, Waugh R, Brown JW, Schulman A, Langridge P, et al. 2012. A physical, genetic and functional sequence assembly of the barley genome. Nature. 491: 711-716.
- International Rice Genome Sequencing Project2005. The map-based sequence of the rice genome. Nature. 436: 793-800.
- International Wheat Genome Sequencing Consortium (IWGSC)2014. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345: 1251788
- Kloosterman B, Abelenda JA, del Gomez MM, Oortwijn M, de Boer JM, Kowitwanich K, et al. 2013. Naturally occurring allele diversity allows potato cultivation in northern latitudes. Nature. 495: 246-250.
- Korlach J, Marks PJ, Cicero RL, Gray JJ, Murphy DL, Roitman DB, et al. 2008. Selective aluminum passivation for targeted immobilization of single DNA polymerase molecules in zero-mode waveguide nanostructures. Proc Natl Acad Sci USA. 105: 1176-1181.
- Lam HM, Xu X, Liu X, Chen W, Yang G, Wong FL, et al. 2010. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet. 42: 1053-1059.
- Li F, Fan G, Wang K, Sun F, Yuan Y, Song G, et al. 2014. Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet. 46: 567-572.
- Li Y, Ruperao P, Edwards D, Batley J, Hobson K, Pang J, et al. 2016. Genome-wide association studies of disease and drought-relative traits using whole genome re-sequencing data in chickpea. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P1018.
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. 2005. Genome sequencing in micro-fabricated high-density picolitre reactors. Nature. 437: 376-380.
- Michael TP, Jackson S. 2013. The first 50 plant genomes. Plant Genome. 6:
- Ouellet T, Gou L, Konkin D, Hattori J, Tekieh F, Wolfe D, et al. 2016. Towards cloning of the Fusarium head blight-resistance locus from Thinopyrum elongatum chromosome 7EL. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0859.
- Pajerowska-Mukhtar K, Stich B, Achenbach U, Ballvora A, Lübeck J, Strahwald J, et al. 2009. Single nucleotide polymorphisms in the allene oxide synthase 2 gene are associated with field resistance to late blight in populations of tetraploid potato cultivars. Genetics. 181: 1115-1127.
- Phillips RL, Vasil IK. 2001. DNA-based markers in plants. 2nd ed. Luwer Academic Publishers. Dordrecht:
- Potato Genome Sequencing ConsortiumXu X, Pan S, Cheng S, Zhang B, Mu D, et al. 2011. Genome sequence and analysis of the tuber crop potato. Nature. 475: 189-195.
- Riedelsheimer C, Czedik-Eysenberg A, Grieder C, Lisec J, Technow F, Sulpice R, et al. 2012. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat Genet. 44: 217-220.
- Ronaghi M, Karamohamed S, Pettersson B, Uhlén M, Nyrén P. 1996. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem. 242: 84-89.
- Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes CA, et al. 1977a. Nucleotide sequence of bacteriophage phi X174 DNA. Nature. 265: 687-695.
- Sanger F, Coulson AR. 1975. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 94: 441-448.
- Sanger F, Nicklen S, Coulson AR. 1977b. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 74: 5463-5467.
- Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, et al. 2010. Genome sequence of the palaeopolyploid soybean. Nature. 463: 178-183.
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. 2009. The B73 maize genome: complexity, diversity, and dynamics. Science. 326: 1112-1115.
- Shaw L, Lv B, Nitcher R, Li C, Han X, Fu D, et al. 2016. Characterization of flowering locus T2 (FT2) in wheat and Brachypodium. In: Proc. of thePlant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0837.
- Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM, et al. 2005. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 309: 1728-1732.
- Shendure JA, Porreca GJ, Church GM. 2008. Overview of DNA sequencing strategies. pp. 7.1.1-7.1.11. John Wiley & Sons.Current protocols in molecular biology. John Wiley & Sons. Bognor Regis, West Sussex.
- Shi Z, Liu S, Noe J, Arelli P, Meksem K, Li Z. 2015. SNP identification and marker assay development for high-throughput selection of soybean cyst nematode resistance. BMC Genomics. 16: 314
- Singh R, Ong-Abdullah M, Low ET, Manaf MA, Rosli R, Nookiah R, et al. 2013. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature. 500: 335-339.
- Sukumaran S, Reynolds M, Cossani M. 2016. Genetic mapping for heat and drought tolerance in spring wheat. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0849.
- Taylor MS, Tornqvist CEI, Grabowski P, Casler M, Jiang Y. 2016. Genetic variation of flowering time and biomass in switchgrass. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0016.
- The Arabidopsis Genome Initiative2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature (London). 408: 796-815.
- Thomelin PML, Bonneau J, Taylor JD, Choulet F, Sourdille P, Langridge P, et al. 2016. Positional cloning of a QTL, qDHY.3BL, on chromosome 3BL for drought and heat tolerance in bread wheat. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0850.
- Tomato Genome Consortium2012. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 485: 635-641.
- Tornqvist CEI, Ramstein GP, Evans J, Vaillancourt B, Crisovan E, Taylor MS, et al. 2016. Genetic analysis of flowering time in switchgrass using a pseudo-F2 population from an upland x lowland cross. In: Proc. of the Plant and Animal Genome Conference (PAGXXIV); San Diego, CA, USA. pp P0015.
- Turcatti G, Romieu A, Fedurco M, Tairi AP. 2008. A new class of cleavable fluorescent nucleotides: synthesis and optimization as reversible terminators for DNA sequencing by synthesis. Nucleic Acids Res. 36: e25
- Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, et al. 2007. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One. 2: e1326
- Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, et al. 2010. The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet. 42: 833-839.
- Vidal RO, do Nascimento LC, Mondego JM, Pereira GA, Carazzolle MF. 2012. Identification of SNPs in RNA-seq data of two cultivars of Glycine max (soybean) differing in drought resistance. Genet Mol Biol. 35(1 Suppl): 331-334.
- Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, et al. Brassica rapa Genome Sequencing Project Consortium2011. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 43: 1035-1039.
- Wu R, Kaiser AD. 1968. Structure and base sequence in the cohesive ends of bacteriophage lambda DNA. J Mol Biol. 35: 523-537.
- Xia Z, Watanabe S, Yamada T, Tsubokura Y, Nakashima H, Zhai H, et al. 2012. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci USA. 109: E2155-E2164.
- Xu J, Yuan Y, Xu Y, Zhang G, Guo X, Wu F, et al. 2014. Identification of candidate genes for drought tolerance by whole-genome resequencing in maize. BMC Plant Biol. 14: 83
- Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science. 296: 79-92.